Complete Guide to CRISPR-Cas9 Gene Knockout in Zebrafish: Protocols, Troubleshooting, and Validation
This comprehensive guide provides researchers and drug development professionals with a detailed, step-by-step protocol for performing CRISPR-Cas9-mediated gene knockout in zebrafish.
Complete Guide to CRISPR-Cas9 Gene Knockout in Zebrafish: Protocols, Troubleshooting, and Validation
Abstract
This comprehensive guide provides researchers and drug development professionals with a detailed, step-by-step protocol for performing CRISPR-Cas9-mediated gene knockout in zebrafish. Covering foundational principles, optimized injection methods, common troubleshooting solutions, and rigorous validation techniques, the article synthesizes current best practices for generating stable knockout lines. It addresses key challenges in efficiency, specificity, and phenotypic analysis, enabling the effective use of zebrafish as a model for functional genomics and preclinical drug discovery.
Understanding CRISPR-Cas9 Zebrafish Knockouts: Principles, Design, and Model Suitability
Why Use Zebrafish for CRISPR-Cas9 Knockout Studies? Advantages Over Other Model Organisms
Within the broader thesis on optimizing CRISPR-Cas9 gene knockout protocols, zebrafish (Danio rerio) have emerged as a preeminent model organism for functional genomics and drug discovery. Their unique combination of biological attributes and experimental tractability offers distinct advantages over mammalian and invertebrate models, making them ideal for high-throughput gene function analysis.
Advantages Over Other Model Organisms
The selection of zebrafish for CRISPR-Cas9 studies is underpinned by several quantifiable benefits.
Table 1: Comparative Analysis of Model Organisms for CRISPR-Cas9 Knockout
| Feature | Zebrafish | Mouse | C. elegans | Fruit Fly |
|---|---|---|---|---|
| Genetic Similarity to Humans | ~70% (82% disease-related genes) | ~85% | ~40% | ~60% |
| Embryonic Development | External, rapid (24-48 hpf for organogenesis) | Internal, slow (~20 days) | Rapid (~3 days) | Rapid (~24 hours) |
| Generation Time | ~3 months | ~3 months | ~3 days | ~2 weeks |
| Offspring per Clutch | 100-300 | 6-12 | ~300 | 50-100 |
| Optical Transparency | Yes (embryos & larvae) | No (except some engineered lines) | Yes | No (in larvae/adult) |
| Ease of CRISPR Delivery | Microinjection into 1-cell embryo | Pronuclear injection; ES cell editing | Microinjection into gonad | Embryo microinjection |
| Husbandry Cost (Relative) | Low | High | Very Low | Low |
| Ethical Regulations | Less stringent (pre-hatch) | Stringent | Minimal | Minimal |
| Suitability for High-Throughput Screening | Excellent | Moderate | Excellent | Good |
- High Throughput & Scalability: Large clutch sizes enable statistical robustness in F0 screening and efficient generation of F1/F2 mutant lines.
- Visualization of Phenotypes: Transparency and external development allow real-time, non-invasive imaging of developmental and cellular processes.
- Conserved Physiology & Pathways: High genetic homology ensures relevance of findings to human biology and disease mechanisms.
- Cost-Effectiveness: Lower maintenance costs and smaller space requirements facilitate large-scale studies.
Application Notes: Key Research Areas
CRISPR-Cas9 knockout in zebrafish is pivotal for modeling human diseases (cancer, cardiovascular, neurological), studying developmental biology, and conducting in vivo drug and toxicology screens.
Detailed Protocols
Protocol 1: Design and Synthesis of CRISPR Components
Objective: To prepare sgRNA and Cas9 mRNA for microinjection.
Materials:
- Target genomic DNA sequence.
- PCR thermocycler.
- T7 RNA polymerase kit.
- SP6 or T7 mMessage mMachine kit.
- RNase-free reagents and equipment.
Method:
- sgRNA Template Preparation: Design a target-specific oligo (20-nt guide + 3-nt NGG PAM upstream). Perform PCR using a T7-promoter-containing forward primer and the target-specific reverse primer.
- In Vitro Transcription (IVT): Purify the PCR product. Use the T7 RNA polymerase kit for IVT. Treat with DNase I. Purify sgRNA using phenol-chloroform extraction or a spin column.
- Cas9 mRNA Synthesis: Linearize a plasmid containing a zebrafish codon-optimized Cas9 cDNA downstream of an SP6/T7 promoter. Use the mMessage mMachine kit for capped mRNA synthesis. Purify as in step 2.
- Quality Control: Quantify RNA concentration via spectrophotometry (260/280 nm ratio ~2.0). Analyze integrity by denaturing agarose gel electrophoresis.
Protocol 2: Microinjection into Zebrafish Embryos
Objective: To deliver CRISPR-Cas9 components into one-cell stage embryos for efficient mutagenesis.
Materials:
- Wild-type AB or TL strain zebrafish.
- Microinjector and micromanipulator.
- Borosilicate glass capillaries.
- Injection mold to create a holding array.
Method:
- Injection Solution: Mix sgRNA (25-50 pg/nl) and Cas9 mRNA (150-300 pg/nl) in nuclease-free water with phenol red tracer.
- Embryo Collection: Set up natural pairwise matings. Collect embryos within 15 minutes post-fertilization.
- Needle Preparation & Loading: Pull capillaries to create fine needles. Back-load 1-2 µL of injection mix.
- Microinjection: Align embryos in grooves on an agarose plate. Using the micromanipulator, pierce the chorion and inject approximately 1 nL of the mix into the cell cytoplasm or yolk near the cell. Target 50-100 embryos per experimental group.
- Post-injection Care: Transfer injected embryos to embryo medium. Incubate at 28.5°C. Remove dead embryos after a few hours.
Protocol 3: Screening for Induced Mutations
Objective: To assess mutagenesis efficiency in injected (F0) and identify germline-transmitted mutations in subsequent generations (F1).
Method – F0 Efficiency (T7 Endonuclease I Assay):
- At 24-48 hpf, pool 10-20 embryos per target. Extract genomic DNA.
- PCR-amplify a ~500-bp region flanking the target site.
- Hybridize and re-anneal PCR products to form heteroduplex DNA if mutations are present.
- Digest with T7E1 enzyme, which cleaves heteroduplex DNA.
- Analyze fragments on an agarose gel. Cleavage bands indicate mutation. Efficiency (%) = (1 - sqrt(1 - (b+c)/(a+b+c))) * 100, where a=uncut band intensity, b & c=cut band intensities.
Method – Germline Screening (Fin Clip):
- Raise injected (F0) fish to adulthood (~3 months).
- Outcross F0 adults to wild-types. At 5-7 dpf, anesthetize and clip a small piece of the caudal fin from the F1 parent for DNA extraction.
- Perform PCR on the fin-clip DNA and sequence the amplicon (Sanger or NGS) to detect specific mutations.
- Raise F1 offspring from founders with confirmed mutations. Screen F1 progeny to identify heterozygous carriers.
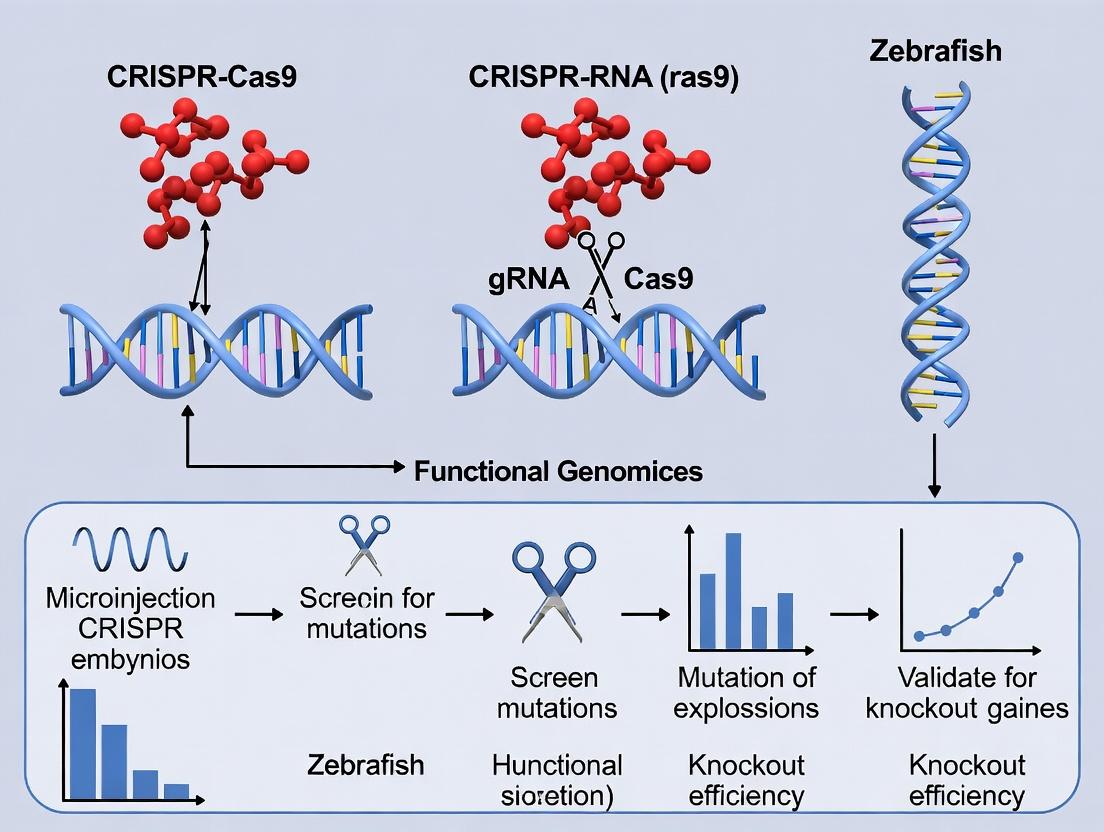
Visualization: Experimental Workflow and Key Pathway
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagent Solutions for Zebrafish CRISPR-Cas9 Experiments
| Item | Function/Application | Example/Notes |
|---|---|---|
| Zebrafish Codon-Optimized Cas9 Plasmid | Source for in vitro transcription of Cas9 mRNA. Ensures high translation efficiency in zebrafish cells. | pT3TS-nCas9n, pCS2-nCas9n. |
| T7 RNA Polymerase Kit | For in vitro transcription of sgRNA from a PCR template containing a T7 promoter. | Commercial IVT kits (e.g., NEB HiScribe). |
| mMessage mMachine Kit (SP6/T7) | For synthesis of capped, polyadenylated Cas9 mRNA to enhance stability and translation. | Thermo Fisher Scientific kits. |
| Phenol Red Solution (0.5%) | Tracer dye added to the injection mix to visualize delivery volume and site. | Non-toxic to embryos. |
| Embryo Medium (E3) | Standard medium for raising zebrafish embryos and larvae. Recipe: 5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl₂, 0.33 mM MgSO₄. | Can be supplemented with methylene blue to inhibit fungal growth. |
| Pronase Solution | Enzyme used to dechorionate (remove the outer membrane) embryos for certain imaging or injection protocols. | 2 mg/mL in E3. |
| Tricaine (MS-222) | Anesthetic for immobilizing larvae or adults for fin clipping, imaging, or sorting. | Stock: 400 mg/mL, pH 7.0. Working: 160 mg/L in E3. |
| Genomic DNA Extraction Buffer | Lysis buffer for rapid preparation of PCR-ready DNA from embryos or fin clips. | Typical components: Tris-HCl, EDTA, Tween-20, Proteinase K. |
| T7 Endonuclease I | Enzyme used to detect indel mutations by cleaving heteroduplex DNA in PCR products. | Key reagent for initial F0 efficiency assessment. |
| Agarose | For preparing injection plates (with molds) and analytical gels for DNA/RNA quality control. | Low-melt agarose for injection plates; standard for gels. |
Application Notes: Core Mechanism and Key Parameters
CRISPR-Cas9 facilitates targeted gene disruption in zebrafish embryos via a two-component system: a single-guide RNA (sgRNA) for target recognition and the Cas9 endonuclease for DNA cleavage. This initiates error-prone non-homologous end joining (NHEJ), leading to insertion/deletion (indel) mutations that can disrupt gene function.
Table 1: Key Quantitative Parameters for CRISPR-Cas9 Targeting in Zebrafish
| Parameter | Typical Range / Value | Impact on Efficiency |
|---|---|---|
| GC Content of sgRNA | 40-60% | Higher specificity & stability |
| sgRNA Length | 20 nucleotides (nt) | Standard for target specificity |
| PAM Sequence (SpCas9) | 5'-NGG-3' | Mandatory 3' adjacent motif |
| Optimal Injection Volume | 1-2 nL per embryo | Minimizes embryo damage |
| Cas9 mRNA Concentration | 100-300 ng/µL | Balance of efficiency & toxicity |
| sgRNA Concentration | 25-100 ng/µL | Must be titrated with Cas9 dose |
| Optimal Injection Time | 1-4 cell stage | Maximizes germline transmission |
| Expected Mutation Rate (F0) | 20-80% (somatic) | Highly target-dependent |
| Germline Transmission Rate | 10-60% in F1 offspring | Requires screening of founders |
Table 2: Common Outcomes and Detection Methods
| Outcome | Molecular Result | Primary Detection Method |
|---|---|---|
| Frame-shift Mutation | Indels not multiples of 3 bp | PCR, Gel Electrophoresis (T7E1 assay) |
| In-frame Mutation | Indels in multiples of 3 bp | Sanger Sequencing |
| Biallelic Disruption (F0) | Mutations in both alleles | High-percentage indels in bulk assay |
| Germline Integration | Mutation passed to F1 | Individual F1 genotyping |
| Off-target Effects | Cleavage at similar sites | Whole-genome sequencing or targeted deep-seq |
Detailed Protocol: Microinjection for Gene Disruption
A. Reagent Preparation
- sgRNA Synthesis: Use target-specific primer with T7 promoter for in vitro transcription. Purify using RNA clean-up kits.
- Cas9 Source: Prepare capped, polyadenylated Cas9 mRNA (or use recombinant protein). Aliquot and store at -80°C.
- Injection Mix: Combine in nuclease-free water:
- Cas9 mRNA (300 ng/µL) OR Cas9 protein (500 ng/µL)
- sgRNA (50 ng/µL)
- Phenol Red (0.1%) for visualization.
- Centrifuge at 14,000 x g for 10 minutes at 4°C before loading needle.
B. Embryo Collection & Injection
- Set up natural zebrafish crosses and collect embryos within 15 minutes post-fertilization.
- Align embryos along a groove in an agarose injection plate.
- Using a microinjector and pulled glass capillary needles, inject 1-2 nL of the mix into the cell yolk or cytoplasm at the 1-4 cell stage.
- Incubate injected embryos in E3 embryo medium at 28.5°C.
C. Screening and Validation (F0 Somatic & F1 Germline)
- At 24-48 hpf: Assess viability and morphology. Pool 8-10 embryos for initial efficiency check.
- Genomic DNA Extraction: Use alkaline lysis or commercial kits.
- Primary PCR: Amplify ~300-500 bp region flanking the target site.
- Mutation Detection:
- T7 Endonuclease I (T7E1) Assay: Denature/anneal PCR products; digest heteroduplex DNA. Analyze on 2% agarose gel.
- Sanger Sequencing: Sequence PCR products directly for trace decomposition analysis, or clone PCR amplicons for individual allele analysis.
- Raise Positive F0 Injected Fish: Screen for germline transmission by outcrossing to wild-type fish and genotyping individual F1 offspring.
Visualization
Title: CRISPR-Cas9 Zebrafish Workflow
Title: Molecular Mechanism of CRISPR-Cas9 Disruption
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for CRISPR-Cas9 in Zebrafish
| Reagent / Material | Function & Purpose | Critical Notes |
|---|---|---|
| T7 RNA Polymerase Kit | In vitro transcription of sgRNA. | Ensures high-yield, capped RNA for stability. |
| Cas9 mRNA (e.g., Spy Cas9) | Encodes the endonuclease. | Optimized for zebrafish codon usage; alternative: recombinant Cas9 protein. |
| Phenol Red (0.1%) | Visual dye for injection mix. | Allows monitoring of injection volume without toxicity. |
| Agarose Injection Molds | Holds embryos for microinjection. | Creates grooves for consistent embryo alignment. |
| Microinjector & Capillaries | Precise delivery of CRISPR components. | Requires calibration for 1-2 nL volumes. |
| T7 Endonuclease I (T7E1) | Detects indels via mismatch cleavage. | Fast, cost-effective initial screening tool. |
| High-Fidelity DNA Polymerase | Amplifies genomic target locus. | Critical for clean PCR before sequencing or T7E1. |
| Gel Extraction Kit | Purifies DNA fragments for sequencing. | Used for cloning PCR products for allele-specific analysis. |
| E3 Embryo Medium | Buffer for embryo incubation. | Maintains osmolarity and health post-injection. |
The success of any CRISPR-Cas9-mediated gene knockout experiment in zebrafish hinges on the initial selection of highly efficient and specific single guide RNAs (sgRNAs). This foundational step determines the rate of mutagenesis, the spectrum of induced mutations, and the potential for off-target effects, directly impacting the reliability of downstream phenotypic analyses. This Application Note details a standardized pipeline for the in silico design and rapid in vivo validation of sgRNAs, forming the critical first chapter of a comprehensive thesis on zebrafish knockout protocols.
sgRNA Design Principles & Quantitative Benchmarks
Effective sgRNA design balances target efficiency with specificity. Key parameters are summarized below.
Table 1: Key Parameters for High-Efficiency sgRNA Design in Zebrafish
| Parameter | Optimal Target/Range | Rationale & Notes |
|---|---|---|
| GC Content | 40-60% | Guides with very low or high GC content show reduced activity. |
| Target Position | Exonic, 5' coding region | Maximizes probability of frameshift and nonsense-mediated decay (NMD). |
| Seed Region (PAM-proximal 8-12 bp) | No secondary structure, high specificity | Critical for R-loop stability and initial Cas9 binding. |
| Predicted Efficiency Score | >60 (Tool-dependent) | Use multiple algorithms (e.g., CRISPOR, CHOPCHOP) for consensus. |
| Off-Target Mismatches | ≥3 mismatches, especially in seed region | Prioritize guides with no highly homologous genomic sequences. |
| PAM Sequence (SpCas9) | 5'-NGG-3' | The canonical SpCas9 PAM; must be present immediately 3' of target. |
Table 2: Comparison of Primary sgRNA Design Tools for Zebrafish
| Tool | Key Features | Zebrafish Genome Support | Output Metrics |
|---|---|---|---|
| CHOPCHOP | User-friendly, visualizes target locus, scores efficiency & off-targets. | Yes (GRCz11) | Efficiency score, off-target count, oligos for cloning. |
| CRISPOR | Integrates multiple scoring algorithms (Doench '16, Moreno-Mateos), detailed off-target analysis. | Yes | MIT & CFD specificity scores, efficiency scores, primer design. |
| UCSC Genome Browser CRISPR Track | Visual design within genomic context, including conservation & chromatin data. | Yes | Primarily visual; requires cross-referencing with other tools. |
Experimental Protocol:In VivoValidation of sgRNA Efficiency
Protocol 1: Rapid F0 ("Crispant") Screening by Fluorescent PCR and Gel Electrophoresis Objective: Qualitatively assess mutagenesis efficiency in injected embryos (F0) prior to raising founders.
Materials & Reagents
- sgRNA/Cas9 Complex: Purified sgRNA (synthesized in vitro or purchased) and recombinant Cas9 protein or Cas9 mRNA.
- Injection Equipment: Microinjector, pulled glass capillary needles, zebrafish embryo injection mold.
- Genomic DNA Extraction Reagents: Lysis buffer (e.g., 10 mM Tris-HCl, pH 8.0, 50 mM KCl, 0.3% Tween-20, 0.3% NP-40, 1 mM EDTA, 1 mg/mL Proteinase K).
- PCR Reagents: High-fidelity DNA polymerase, primers flanking the target site (~300-500 bp amplicon).
- Gel Electrophoresis: Standard agarose gel (2-4%) or high-resolution system (e.g., QIAxcel Advanced, LabChip GX).
Methodology
- sgRNA Preparation: Synthesize sgRNA via in vitro transcription (IVT) from a dsDNA template or purchase as synthetic, chemically-modified RNA.
- Microinjection: Prepare injection mix (final concentration: ~25-50 pg nL⁻¹ Cas9 protein or 100-300 pg nL⁻¹ Cas9 mRNA + 25-100 pg nL⁻¹ sgRNA). Inject 1-2 nL into the cell cytoplasm of 1-4 cell stage zebrafish embryos.
- Sample Collection: At 24-48 hours post-fertilization (hpf), pool 8-10 injected embryos and 5 control embryos. Homogenize individually in 50 µL lysis buffer and incubate at 55°C for 2 hours, followed by 95°C for 10 minutes to inactivate Proteinase K.
- PCR Amplification: Perform PCR using 1-2 µL of crude lysate as template. Include a non-injected control.
- Heteroduplex Analysis:
- For standard agarose gel: Denature and reanneal PCR products (95°C for 5 min, ramp down to 25°C at -2°C/sec). Run on a 2-4% agarose gel. A diffuse/smeared band above the WT amplicon indicates heterogeneous indels.
- For high-resolution systems: Follow manufacturer's protocol. Cleavage traces will show multiple peaks downstream of a clean WT peak.
Protocol 2: Quantitative Validation by Next-Generation Sequencing (NGS) Objective: Precisely quantify indel percentage and spectrum for candidate sgRNAs.
Methodology
- Amplicon Library Preparation: Perform PCR from individual or pooled embryo lysates (as above) using primers with overhangs containing Illumina adapter sequences.
- Indexing PCR: Add unique dual indices (i5 and i7) via a second, limited-cycle PCR.
- Sequencing: Pool libraries and run on a MiSeq or similar platform (2x250 bp recommended).
- Data Analysis: Use tools like CRISPResso2 or ICE (Inference of CRISPR Edits). Input demultiplexed FASTQ files, the target amplicon sequence, and the sgRNA sequence. Output includes: % Indel, indel size distribution, and precise sequence alleles.
Visualization of Workflows & Pathways
Title: sgRNA Design and Validation Workflow for Zebrafish
Title: DNA Repair Pathways After CRISPR-Cas9 Cleavage
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for sgRNA Validation in Zebrafish
| Item | Function & Rationale | Example/Notes |
|---|---|---|
| Recombinant Cas9 Protein | Immediate activity; no transcription/translation delay. Reduces mosaicism. | GeneArt Platinum Cas9, TrueCut Cas9 Protein. |
| Chemically Modified sgRNA | Enhanced nuclease resistance, improved stability in vivo, higher efficiency. | Synthego sgRNA EZ Kit, Trilink CleanCap sgRNA. |
| High-Sensitivity DNA Analysis System | Quantitative, capillary electrophoresis-based analysis of PCR amplicons for indel efficiency. | PerkinElmer LabChip GX, QIAGEN QIAxcel Advanced. |
| CRISPR Analysis Software (NGS) | Accurately quantifies editing efficiency and characterizes mutation spectra from NGS data. | CRISPResso2 (open source), ICE (Synthego), BE-Analyzer (for base editors). |
| Zebrafish Embryo Lysis Buffer | Rapid, single-tube genomic DNA preparation from pooled embryos for PCR. | 10mM Tris-Cl, 50mM KCl, 0.3% Tween-20, 0.3% NP-40, 1mg/mL Proteinase K. |
| Next-Generation Sequencing Kit | For precise, quantitative validation of sgRNA efficiency and off-target profiling. | Illumina MiSeq Reagent Kit v2 (500 cycles). |
This application note is framed within a thesis on CRISPR-Cas9 gene knockout protocols in zebrafish (Danio rerio) research. The choice of Cas9 protein variant and its delivery format is critical for experimental success, influencing mutation efficiency, specificity, and phenotypic outcomes. This document provides a comparative analysis and detailed protocols to guide researchers and drug development professionals in selecting and implementing the optimal Cas9 system.
Cas9 Variants: Characteristics and Applications
The primary variants of Streptococcus pyogenes Cas9 (SpCas9) differ in their nuclease activity, dictating their application for specific genome engineering goals.
Table 1: Comparison of Cas9 Protein Variants for Zebrafish Gene Knockout
| Cas9 Variant | Nuclease Domains Active | DNA Cleavage Outcome | Primary Application in Zebrafish | Typical Mutation Efficiency (Indel %) | Key Advantage |
|---|---|---|---|---|---|
| Wild-type (wtCas9) | RuvC & HNH | Double-Strand Break (DSB) | Complete gene knockout via NHEJ. | 50-90% (F0 screening) | Highest efficiency for generating frameshifts. |
| Nickase (Cas9n/D10A) | HNH only | Single-Strand Break ("Nick") | Paired nicks for reduced off-targets. | 30-70% (paired guides) | Dramatically lower off-target mutagenesis. |
| Dead Cas9 (dCas9) | None | No cleavage, DNA binding only. | Transcriptional repression (CRISPRi), imaging, base editing fusions. | N/A (no cleavage) | Enables knock-down without genetic mutation. |
Delivery Formats: Considerations for Zebrafish
Cas9 protein must be delivered into the zebrafish embryo, typically at the one-cell stage. The format impacts stability, activity window, and mosaicism.
Table 2: Comparison of Cas9 Delivery Formats in Zebrafish
| Delivery Format | Components Delivered | Preparation Complexity | Activity Onset | Duration of Activity | Best For |
|---|---|---|---|---|---|
| Cas9-sgRNA RNP (Ribonucleoprotein) | Pre-complexed purified Cas9 protein + sgRNA | Moderate (protein purification/commercial) | Immediate (<30 min) | Short (hours) | Rapid degradation, minimal off-targets, high F0 knockouts. |
| Cas9 mRNA + sgRNA | In vitro transcribed mRNA + sgRNA | High (multiple IVT steps) | Delayed (1-3 hrs) | Moderate (up to 24 hrs) | Sustained expression, higher mosaicism in F0. |
| Plasmid DNA | Expression vector(s) for Cas9 + sgRNA | Low (standard cloning) | Significantly delayed (>6 hrs) | Prolonged (days) | Germline transmission studies, not recommended for efficient F0 knockout. |
Detailed Protocols
Protocol 1: Gene Knockout using Cas9 Protein RNP Complexes
This is the preferred method for high-efficiency, low off-target F0 knockout screening.
Materials (Research Reagent Solutions):
- Purified Cas9 Protein (wt or variant): Commercial source (e.g., Thermo Fisher TrueCut Cas9, IDT Alt-R S.p. Cas9) or lab-purified. Functional core reagent.
- Target-specific sgRNA: Chemically synthesized (IDT Alt-R) or in vitro transcribed from a DNA template. Defines targeting specificity.
- Microinjection Buffer (1x): Typically 10 mM Tris-HCl, 0.1 mM EDTA, pH 7.5. Maintains RNP stability during injection.
- Phenol Red (0.1%): Visual tracer for microinjection. Non-functional aid.
- Zebrafish Embryos: One-cell stage, collected within 15 minutes post-fertilization (mpf).
Methodology:
- sgRNA Preparation: Resynthesize or dilute sgRNA in nuclease-free microinjection buffer to a working concentration of 100-200 ng/µL.
- RNP Complex Formation: Combine the following in a tube:
- 2 µL Cas9 protein (final conc. ~300-500 ng/µL)
- 2 µL sgRNA (final conc. ~50-100 ng/µL)
- 1 µL Phenol Red Incubate at 37°C for 10 minutes to allow RNP complex assembly.
- Microinjection: Load the RNP mix into a needle and inject approximately 1 nL (~200-500 pg of Cas9 protein) into the cytoplasm of a one-cell stage zebrafish embryo.
- Embryo Handling: Incubate injected embryos at 28.5°C in E3 embryo medium. Screen for mutagenesis at 24-48 hours post-fertilization (hpf) via PCR/restriction enzyme (RE) assay or later via T7 Endonuclease I (T7EI) assay on pooled embryos.
Protocol 2: Paired Nicking for High-Fidelity Knockouts
Uses two Cas9n (D10A) proteins with offset sgRNAs to create staggered DSBs, improving specificity.
Methodology:
- sgRNA Design: Design two sgRNAs targeting opposite DNA strands of the target locus with a 5-50 bp offset.
- RNP Formation: Form separate RNP complexes for each sgRNA with Cas9n protein as in Protocol 1, Step 2.
- Complex Mixing: Combine equal volumes of the two RNP complexes immediately before injection.
- Microinjection & Analysis: Inject the mixed RNPs as in Protocol 1, Steps 3-4. Analyze cleavage efficiency. Note that indels are only produced when both nicks occur, resulting in higher specificity but potentially lower overall efficiency.
Visualizing Selection and Workflow
Title: Decision Tree for Selecting Cas9 Variant and Delivery Format
Title: Workflow for Zebrafish Gene Editing using Cas9 RNP Complexes
Effective pre-injection planning is the critical foundation for successful CRISPR-Cas9-mediated gene knockout in zebrafish. Within the broader thesis on optimizing these protocols, this phase dictates experimental validity, reproducibility, and ethical compliance. This document outlines a standardized timeline, enumerates essential ethical considerations, and defines the required experimental and procedural controls.
Detailed Pre-injection Timeline
The pre-injection phase encompasses a 4-6 week period prior to microinjection day. Adherence to this timeline ensures optimal germline transmission rates and animal welfare.
Table 1: Pre-injection Planning Timeline
| Week | Activity | Key Deliverables & Notes |
|---|---|---|
| -6 to -4 | Target Selection & gRNA Design | In silico analysis for on/off-target scores; Design of 2-3 gRNAs per target. |
| -4 | Ethical & Biosafety Approval | Submission of IACUC and IBC protocols; Approval mandatory before proceeding. |
| -4 | gRNA Synthesis & Validation | Synthesis via in vitro transcription; Validate quality via gel electrophoresis. |
| -3 | Cas9 Protein/RNA Preparation | Acquire high-quality Cas9 nuclease (protein or mRNA). Aliquot and store at -80°C. |
| -2 | Setup of Breeding Tanks | Acclimate wild-type adult zebrafish pairs in dedicated breeding systems. |
| -1 | Injection Setup Validation | Calibrate microinjector and needles using dye; practice embryo handling. |
| Day -1 | Final Preparation | Prepare final injection mixes; set up embryo collection apparatus. |
Ethical Considerations and Regulatory Compliance
Zebrafish are vertebrate models subject to ethical oversight. Key considerations include:
The 3Rs Principle (Replace, Reduce, Refine):
- Replace: Use in silico models for preliminary gRNA design. Justify animal use relative to research goals.
- Reduce: Optimize injection mixes and techniques to minimize the number of embryos injected. Use power analysis to determine minimal necessary sample size (N).
- Refine: Use anesthesia (e.g., Tricaine) for embryo handling post-injection. Define and implement humane endpoints for severe morphological defects.
Germline Modification: Research must be classified as basic, non-applicative research. Any protocol intending to create stable, heritable lines must have explicit IACUC approval. Embryos should not be raised beyond 120 hours post-fertilization (hpf) if exhibiting severe defects.
Data Management and Transparency: All experimental parameters (gRNA sequences, concentrations, numbers of embryos) must be meticulously recorded. Negative and off-target data must be archived.
Biosafety (NIH Guidelines): CRISPR-Cas9 experiments are generally considered Biosafety Level 1 (BSL-1). However, IBC review is required to confirm containment practices.
Required Experimental and Procedural Controls
Defined controls are non-negotiable for interpreting knockout efficacy and specificity.
Table 2: Mandatory Experimental Controls
| Control Type | Purpose | Protocol Implementation |
|---|---|---|
| Uninjected Control | Baseline for normal development and genotyping background. | Raise a clutch of embryos from the same breeding pair without manipulation. |
| Standard Control (Cas9-only) | Identifies effects due to Cas9 toxicity or injection trauma. | Inject embryos with nuclease (Cas9 protein/mRNA) alone, without gRNA. |
| gRNA-only Control | Assesses potential toxicity of the gRNA itself. | Inject embryos with gRNA complexed with vehicle, but without active Cas9. |
| Targeting Control (e.g., tyr) | Validates the entire injection system is functional. | Co-inject a well-characterized gRNA (e.g., for tyrosinase, causing albinism) with Cas9 as a positive control for mutagenesis. |
| Non-targeting gRNA Control | Control for non-specific gRNA effects. | Inject with Cas9 and a scrambled gRNA sequence with no known genomic target. |
| Replication Control | Ensures reproducibility. | All injections must be performed in at least three independent biological replicates (separate clutches on different days). |
Detailed Protocols for Key Pre-injection Experiments
Protocol 5.1: In Vitro gRNA Synthesis and Validation
- Template Preparation: Amplify gRNA template via PCR using a primer containing the T7 promoter and target sequence.
- In Vitro Transcription (IVT): Use the T7 High-Yield RNA Synthesis Kit. Assemble reaction: 1µg template DNA, 1x buffer, 1x NTPs, 1x T7 RNA polymerase. Incubate 4 hours at 37°C.
- DNase I Treatment: Add 1µL DNase I, incubate 15 min at 37°C.
- Purification: Use RNA clean-up kit. Elute in nuclease-free water.
- Validation: Run 100ng on a 2% agarose/TAE gel. A single, sharp band at ~100-150bp confirms integrity.
Protocol 5.2: Injection Mix Preparation and Calibration
- Master Mix (for 1x concentration):
- Nuclease-Free Water: variable volume
- 10x Injection Buffer (1 mM Tris, 0.1 mM EDTA, pH 7.5): 1 µL
- Phenol Red (0.5%): 0.5 µL (tracer dye)
- Cas9 Protein (e.g., 1 µg/µL): 1 µL (Final: ~100-200 pg per embryo)
- gRNA (e.g., 100 ng/µL): 1 µL (Final: ~20-50 pg per embryo)
- Final Volume: 10 µL
- Calibration: Load mix into a pulled glass capillary needle. Inject a drop of mineral oil onto a hemocytometer. Adjust injection pressure and duration until droplet diameter is ~0.2-0.5 nL (calibrated via dye dilution).
Visualizations
Pre-injection Timeline Workflow (6 weeks to Day 0)
Hierarchy of Mandatory Experimental Controls
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Pre-injection Planning
| Item | Function & Rationale | Example Product/Note |
|---|---|---|
| CRISPR-Cas9 Design Software | Identifies high-efficiency, specific gRNA sequences; predicts off-target sites. | CHOPCHOP, CRISPRscan, IDT's design tool. |
| T7 High-Yield RNA Synthesis Kit | Robust, standardized in vitro transcription for high-yield gRNA synthesis. | NEB HiScribe T7 Kit. |
| Nuclease-free Duplex Buffer | For complexing gRNA with Cas9 protein to form active ribonucleoprotein (RNP). | IDT Duplex Buffer or equivalent. |
| Recombinant Cas9 Nuclease | High-purity, ready-to-use nuclease. Protein form reduces mosaicism vs. mRNA. | GeneArt Platinum Cas9 Nuclease. |
| Phenol Red (0.5%) | Non-toxic injection tracer dye. Allows visual confirmation of delivery. | Diluted in 1x injection buffer. |
| Microinjector & Micromanipulator | Precise, reproducible pneumatic delivery of nL volumes into embryos. | Pneumatic PicoPump (PV820) with manipulator. |
| Borosilicate Glass Capillaries | For pulling fine, sharp injection needles. | 1.0 mm OD, 0.78 mm ID, with filament. |
| Tricaine (MS-222) | Anesthetic for humane handling of embryos post-injection. | Stock: 400 mg/mL, pH 7.0; Use at 160 mg/L. |
Step-by-Step CRISPR-Cas9 Knockout Protocol: From Microinjection to Founder Screening
Application Notes
This protocol details the in vitro preparation of the sgRNA:Cas9 ribonucleoprotein (RNP) complex, the preferred method for CRISPR-Cas9-mediated gene knockout in zebrafish embryos. Direct RNP microinjection offers high efficiency, rapid action, and reduced off-target effects compared to DNA-based methods. It minimizes mosaicism in F0 founders and accelerates functional genetic analysis. The preparation of a high-activity, nuclease-free RNP complex is the critical first step for successful mutagenesis.
Detailed Protocol
Materials
- Research Reagent Solutions
- Cas9 Nuclease: High-specificity, carrier-free protein (e.g., S. pyogenes Cas9). Function: The DNA endonuclease that performs the double-strand break.
- sgRNA (single-guide RNA): Chemically synthesized or in vitro transcribed (IVT). Function: Combines crRNA and tracrRNA to guide Cas9 to the specific genomic target sequence.
- Nuclease-Free Duplex Buffer (e.g., 30 mM HEPES, 100 mM potassium acetate): Function: Optimized ionic environment for RNP complex formation and stability.
- Nuclease-Free Water: Function: To dilute reagents and avoid degradation of RNA/protein.
- Phenol Red Solution (0.5%): Optional additive for microinjection mixes. Function: Visual tracking dye for injection.
Methodology
Step 1: Design and Acquisition of sgRNA
- Identify a 20-nt target sequence (protospacer) directly upstream of a 5'-NGG-3' Protospacer Adjacent Motif (PAM) in your zebrafish gene of interest.
- Prioritize target sites with high on-target and low off-target prediction scores using design tools (e.g., CHOPCHOP, CRISPOR).
- Order the sgRNA as a synthetic, chemically modified RNA (recommended for highest consistency) or as a DNA template for in vitro transcription.
Step 2: In Vitro Transcription of sgRNA (if using IVT method)
- Prepare a DNA template via PCR using a primer containing the T7 promoter sequence.
- Set up the transcription reaction using a T7 RNA polymerase kit.
- Template DNA: 200-500 ng
- NTPs: 7.5 mM each
- T7 Reaction Buffer: 1X
- T7 RNA Polymerase: 0.5 µL
- Nuclease-free water to 10 µL
- Incubate at 37°C for 2-4 hours.
- Add 1 µL of DNase I (RNase-free) and incubate at 37°C for 15 minutes to digest the DNA template.
- Purify the sgRNA using a spin-column-based RNA clean-up kit. Elute in nuclease-free duplex buffer.
- Quantify concentration (ng/µL) via spectrophotometry and assess integrity via denaturing agarose gel electrophoresis.
Step 3: RNP Complex Assembly
- Thaw and prepare all components on ice.
- Dilute sgRNA: Dilute purified or resuspended synthetic sgRNA to a working stock of 100 µM in nuclease-free duplex buffer.
- Prepare the complex mixture in a low-protein-binding microcentrifuge tube:
- Cas9 Protein: 1 µL of 60 µM stock (final: 6 µM)
- sgRNA: 1 µL of 100 µM stock (final: 10 µM)
- Duplex Buffer: 8 µL
- Final Volume: 10 µL
Key: A 1:1.7 molar ratio of Cas9:sgRNA is used to ensure all Cas9 is complexed while minimizing unbound sgRNA.
- Mix gently by pipetting. Do not vortex.
- Incubate at 37°C for 10 minutes to allow proper complex folding, then immediately place on ice. The RNP complex is now ready for injection or can be stored at -80°C for short periods.
Step 4: Microinjection Mix Preparation (Typical Example)
- Immediately before injection, prepare the injection mix on ice:
- Assembled RNP Complex: 2.0 µL
- Phenol Red (0.5%): 0.3 µL
- Nuclease-Free Water: 0.7 µL
- Final Injection Volume: 3.0 µL
- Mix gently by pipetting. Centrifuge briefly to collect contents.
- Load into a microinjection needle. Inject 1-2 nL per embryo at the 1-cell stage.
Data Presentation
Table 1: Recommended Reagent Concentrations for RNP Assembly
| Component | Stock Concentration | Volume in 10 µL Assembly | Final Concentration in Assembly | Purpose |
|---|---|---|---|---|
| Cas9 Nuclease | 60 µM | 1.0 µL | 6.0 µM | DNA cleavage enzyme |
| sgRNA | 100 µM | 1.0 µL | 10.0 µM | Target sequence guide |
| Duplex Buffer | 1X | 8.0 µL | 1X | Optimal complex formation |
Table 2: Troubleshooting Common RNP Preparation Issues
| Problem | Potential Cause | Solution |
|---|---|---|
| Low mutagenesis efficiency | sgRNA degradation | Use synthetic, chemically modified sgRNA; ensure nuclease-free conditions. |
| Incorrect Cas9:sgRNA ratio | Titrate ratio from 1:1 to 1:2 (Cas9:sgRNA). Verify protein/RNA quantification. | |
| Poor sgRNA design | Re-design sgRNA with updated algorithm scores; check for secondary structure. | |
| Embryo toxicity | Excessive injection volume/RNP concentration | Reduce injection volume to ≤2 nL; dilute RNP mix 1.5-2 fold. |
| Unstable injection mix | Lack of carrier/salt | Include 0.1% Phenol Red and ensure Duplex Buffer contains KCl/NaCl. |
Visualizations
Title: RNP Complex Prep & Injection Workflow
Title: Three-Step RNP Assembly Process
Thesis Context
This document provides detailed application notes for the microinjection setup phase of a CRISPR-Cas9 gene knockout workflow in zebrafish (Danio rerio). The protocols herein are critical for achieving high-efficiency mutagenesis and subsequent phenotypic analysis in developmental genetics and drug discovery research.
Microinjection Needle Pulling and Preparation
Detailed Protocol
- Pipette Selection: Use borosilicate glass capillaries with an outer diameter of 1.0 mm and an inner diameter of 0.78 mm, containing an internal filament for back-filling.
- Puller Setup: Program a multi-step pull on a programmable pipette puller (e.g., Sutter P-97/1000). Critical parameters vary by puller model and filament type.
- Pulling Parameters (Sutter P-97, Box Filament):
- Heat: 500
- Pull: 60
- Velocity: 60
- Time: 200
- Pressure: 500
- Use a two-line program. The settings must be empirically optimized for each puller and filament batch.
- Needle Evaluation: Under a high-magnification stereomicroscope, assess the needle for a long, gradual taper and a fine, closed tip. The ideal tip opening, post-breaking, should be <5 µm.
- Tip Breaking: Using fine forceps under a microscope, gently break the very tip of the needle at a 30-45° angle to create a sharp, open point for piercing the chorion.
Table 1: Optimized Pipette Puller Parameters for Common Instruments
| Puller Model | Filament Type | Heat | Pull | Velocity | Time | Pressure | Resulting Tip ID* |
|---|---|---|---|---|---|---|---|
| Sutter P-97 | Box | 500 | 60 | 60 | 200 | 500 | < 5 µm |
| Sutter P-2000 | - | 300 | 40 | 90 | 220 | 500 | < 3 µm |
| Narishige PN-31 | - | 70.5 | - | - | - | - | 3-7 µm |
*Internal Diameter after breaking. All parameters are starting points and require validation.
Injection Needle Calibration and Setup
Detailed Protocol
- System Assembly: Attach the pulled needle to the needle holder of a micromanipulator connected to a pneumatic microinjector (e.g., Picospritzer III) or a syringe pump system.
- Back-Filling: Using a fine, elongated microloader tip, back-fill the needle with ~2 µL of injection solution (e.g., Cas9 protein + sgRNA mix, or reconstituted ribonucleoprotein complex). Avoid bubbles.
- Pressure & Timing Calibration:
- Place a drop of mineral oil on a micrometer slide.
- Immerse the needle tip and depress the foot pedal to inject into the oil.
- Measure the diameter of the resulting spherical droplet. Calculate volume using V = (4/3)πr³.
- Adjust injection pressure (typical range: 10-30 psi) and pulse duration (typical range: 10-100 ms) until the desired injection volume is achieved.
- Target Volume: For 1-4 cell stage zebrafish embryos, the target injection volume is typically 1-2 nL. Exceeding this can cause high embryo mortality.
Table 2: Microinjection Calibration Guide for Zebrafish Embryos
| Target Volume (nL) | Approx. Droplet Diameter in Oil (µm) | Typical Pressure (psi) | Typical Pulse Duration (ms) | Embryo Stage |
|---|---|---|---|---|
| 0.5 | 98 | 8-12 | 10-20 | Shield, etc. |
| 1.0 | 124 | 12-18 | 20-40 | 1-cell (optimal) |
| 1.5 | 142 | 15-22 | 30-50 | 1-4 cell |
| 2.0 | 156 | 18-30 | 40-70 | 1-4 cell |
| >2.5 | >168 | >25 | >70 | Toxic, high mortality |
Embryo Orientation and Injection
Detailed Protocol
- Preparation: Post-fertilization, dechorionate embryos if necessary using pronase treatment or fine forceps. Align embryos in rows on an agarose injection mold (1.5-2% agarose in E3 embryo medium) with grooves sized to hold embryos.
- Orientation: Using a hair loop or fine brush, orient 1-cell stage embryos so the cell is facing the needle and the yolk is opposite. The goal is to inject directly into the cell cytoplasm or the yolk cell just below the blastodisc.
- Injection Technique:
- Lower the needle at a 10-30° angle relative to the plate surface.
- Pierce the chorion (if present) and the cell membrane in one smooth motion.
- Depress the foot pedal to deliver the injection volume.
- Swiftly retract the needle. A successful cytoplasmic injection shows slight displacement of cytoplasmic granules.
- Post-Injection Care: Immediately transfer injected embryos to fresh E3 medium. Incubate at 28.5°C. Remove dead or unfertilized embryos within a few hours.
Workflow Diagram
Diagram Title: Zebrafish CRISPR Microinjection Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Zebrafish CRISPR Microinjection
| Item | Function & Rationale | Example Product/Catalog |
|---|---|---|
| Borosilicate Capillaries w/ Filament | Glass needles for injection. Internal filament enables reliable back-filling by capillary action. | Sutter BF-100-78-10 |
| Programmable Pipette Puller | Produces consistent, fine-tipped needles with customizable taper. | Sutter P-97 Flaming/Brown |
| Pneumatic Microinjector | Delivers precise, pressure-driven pulses of injection mixture. | Parker Picospritzer III |
| Micromanipulator | Allows stable, precise 3D positioning of the injection needle. | Narishige M-152 |
| Agarose (Low Melt) | For creating injection molds to hold and orient embryos. | Fisher BioReagents BP165-25 |
| Phenol Red (0.5%) | Visual aid tracer added to injection mix to confirm delivery. | Sigma P0290 |
| Microloader Pipette Tips | For back-filling needles without introducing bubbles. | Eppendorf 5242956003 |
| Embryo Medium (E3) | Isotonic medium for maintaining embryos pre- and post-injection. | 5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl₂, 0.33 mM MgSO₄ |
| Gene-specific sgRNA | Target-specific component of the CRISPR-Cas9 ribonucleoprotein complex. | Synthesized in vitro or commercially designed. |
| Recombinant Cas9 Protein | Endonuclease that creates double-strand breaks at the sgRNA-targeted locus. | GeneArt Platinum Cas9 Nuclease |
This application note details the critical parameters for the successful microinjection of CRISPR-Cas9 components into zebrafish embryos to achieve efficient gene knockout. As part of a comprehensive thesis on CRISPR-Cas9 protocols in zebrafish, this section focuses on the precise execution of the injection itself, which directly determines mutagenesis efficiency and embryo viability. Optimal dosage, precise developmental timing, and stringent maintenance of embryo health are interdependent factors that must be rigorously controlled.
Dosage Optimization for CRISPR-Cas9 Components
The concentration of Cas9 protein and guide RNA (gRNA) is the primary determinant of both on-target mutagenesis efficiency and off-target effects. Recent studies emphasize a balanced ratio to maximize knockouts while minimizing toxicity.
Table 1: Recommended Dosage Ranges for CRISPR-Cas9 Microinjection in Zebrafish
| Component | Concentration Range | Typical Volume Injected (nL) | Final Amount per Embryo | Key Consideration |
|---|---|---|---|---|
| Cas9 mRNA | 100 - 300 ng/µL | 1 - 2 nL | 100 - 600 pg | Higher concentrations increase efficiency but also toxicity and mosaicism. |
| Cas9 Protein (RNP) | 20 - 60 µM (≈ 300 - 900 ng/µL) | 1 - 2 nL | 300 - 1800 pg | More efficient and faster than mRNA; lower dosage required. |
| sgRNA | 25 - 100 ng/µL | 1 - 2 nL | 25 - 200 pg | Must be titrated with Cas9 concentration. A 1:1 to 1:2 molar ratio (Cas9:gRNA) is often optimal. |
| Phenol Red Tracer | 0.05 - 0.1% | N/A | N/A | Added to injection mix for visualization; non-toxic at this concentration. |
Note: The final injection mix typically combines Cas9 (mRNA or protein) and sgRNA in nuclease-free water or a mild buffer (e.g., 10 mM Tris, 0.25 mM EDTA, pH 7.4). The inclusion of phenol red at 0.05% allows visualization of successful cytoplasmic delivery.
Protocol: Preparing and Titrating the Injection Mix
Objective: To prepare a series of Cas9 RNP concentrations for empirical determination of the optimal dosage for a specific target.
Materials:
- Purified recombinant Cas9 protein (e.g., S. pyogenes)
- Target-specific sgRNA (chemically synthesized or in vitro transcribed and purified)
- Nuclease-free water
- Phenol Red (0.5% stock)
- Microcentrifuge tubes
- Microloader tips
Methodology:
- Prepare Stock Solutions: Dilute Cas9 protein to 60 µM and sgRNA to 200 ng/µL in nuclease-free water.
- Prepare RNP Complex: Pre-complex Cas9 and sgRNA at the desired molar ratio (e.g., 1:1.5) by mixing components in a tube. Incubate at 25°C for 10 minutes to allow RNP formation.
- Prepare Injection Series: Create a dilution series of the pre-complexed RNP in nuclease-free water. A typical series might be: 60 µM, 40 µM, 20 µM, and 10 µM final Cas9 concentration.
- Add Tracer: To each dilution, add Phenol Red stock to a final concentration of 0.05-0.1%. Mix gently by pipetting.
- Centrifuge: Briefly spin down tubes at >10,000 x g for 1 minute to remove bubbles.
- Loading: Using a microloader tip, carefully back-fill a pulled glass needle with the injection mix. Avoid introducing air bubbles.
- Test Injection: Inject approximately 50-100 embryos per concentration. Assess immediate survival and morphology at 2-4 hours post-injection (hpi).
- Efficiency Assessment: At 24-48 hours post-fertilization (hpf), collect 10-20 embryos per group for genomic DNA extraction and PCR/restriction enzyme (RE) assay or T7 Endonuclease I (T7EI) assay to assess indel formation efficiency. The concentration yielding >70% mutagenesis with >80% embryo survival at 24 hpf is optimal.
Timing of Microinjection
Injection must be performed during the single-cell to early cleavage stages to ensure the CRISPR components are incorporated into as many blastomeres as possible, reducing mosaicism in the resulting F0 generation.
Table 2: Developmental Stages for Optimal Microinjection
| Stage | Time Post-Fertilization | Morphological Cues | Rationale & Outcome |
|---|---|---|---|
| Single Cell | 0 - 15 minutes | Embryo is a uniform sphere; cell boundary not yet visible. | Maximizes distribution to all cells, minimizing mosaicism. Technically challenging due to chorion strength. |
| 1-4 Cell | 15 - 45 minutes | Clear cleavage divisions visible. | Ideal practical window. Cytoplasm is accessible, and components disperse into dividing cells. |
| >8 Cell | >1 hour | Multiple small blastomeres. | Increases risk of high mosaicism, as components may not reach all progenitor cells. |
Protocol: Synchronizing Embryo Collection and Injection
- Setup Mating Tanks: Place zebrafish breeding pairs in divided tanks the afternoon before injection.
- Initiate Spawning: At lights-on, remove dividers. Collect embryos immediately upon spawning (within 10-15 minutes).
- Dechorionation (Optional but Recommended): Treat embryos with 1-2 mg/mL pronase in system water for 5-10 minutes. Gently swirl and wash 3x with clean system water. This softens the chorion for easier needle penetration.
- Alignment: Using a fine brush or pipette, align 50-100 dechorionated embryos along the groove of an injection mold (e.g., 2% agarose plate) submerged in injection solution (e.g., 0.3x Danieau's solution).
- Rapid Injection: Complete injection of all embryos within 45 minutes post-fertilization. Prioritize visibly dividing (1-4 cell) embryos.
Monitoring and Maintaining Embryo Health
Post-injection care is vital for survival to screening stages. Stressors include mechanical damage, osmotic shock, and bacterial/fungal infection.
Key Health Assessment Metrics
Table 3: Embryo Health Assessment Post-Microinjection
| Time Point | Normal Phenotype | Signs of Toxicity/Stress | Corrective Action |
|---|---|---|---|
| 1-4 hpi | Clear cytoplasm, intact yolk, symmetrical cells. | Blebbing, lysis, gross asymmetry, cloudy cytoplasm. | Review dosage; ensure injection solution is isotonic (e.g., 0.3x Danieau's). |
| 24 hpf | Normal epiboly, shield formation; no delay. | Developmental delay, necrosis, abnormal morphology. | Reduce Cas9/gRNA concentration; check needle calibration to minimize volume. |
| 48 hpf | Regular somites, beating heart, tail detachment. | Pericardial edema, shortened body axis, lack of circulation. | Improve water quality (frequent changes); add antifungal agent (e.g., Methylene Blue, 0.0001%). |
Protocol: Post-Injection Embryo Care and Screening
- Post-Injection Recovery: Immediately after injection, gently transfer embryos from the injection plate to a clean Petri dish with fresh E3 embryo medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl₂, 0.33 mM MgSO₄) or system water.
- Incubation: Maintain embryos in an incubator at 28.5°C. Do not exceed 50 embryos per 60 mm dish.
- Daily Maintenance:
- Remove any dead or necrotic embryos promptly.
- Change 80-90% of the medium daily using a fine pipette.
- From 24 hpf onward, add PTU (1-Phenyl-2-thiourea, 0.003%) to the medium to inhibit pigment formation if needed for imaging.
- Health Screening: At 24 hpf, sort embryos under a stereomicroscope. Discard those with severe developmental defects. Only apparently healthy embryos should be raised for subsequent genotyping.
Title: CRISPR Microinjection and Embryo Screening Workflow
The Scientist's Toolkit: Key Reagent Solutions
Table 4: Essential Research Reagents for Zebrafish CRISPR Microinjection
| Reagent/Solution | Function & Rationale | Example/Formulation |
|---|---|---|
| Recombinant Cas9 Protein | Direct delivery of nuclease activity; faster, more efficient, and potentially less toxic than mRNA. Enables RNP complex formation. | Commercially available S. pyogenes Cas9, HPLC-purified. |
| Target-Specific sgRNA | Guides Cas9 to the genomic locus of interest. Chemical synthesis ensures high purity and reduces immune response triggers. | Synthesized with 2'-O-methyl 3' phosphorothioate modifications at 3-terminal bases for stability. |
| Phenol Red (0.05-0.1%) | A visually harmless dye used as a tracer in the injection mix to confirm successful cytoplasmic delivery and approximate volume. | Diluted from 0.5% stock in nuclease-free water. |
| Danieau's Solution (0.3x) | An isotonic solution used for holding embryos during injection; minimizes osmotic shock. | 19.3 mM NaCl, 0.23 mM KCl, 0.13 mM MgSO₄, 0.2 mM Ca(NO₃)₂, 1.7 mM HEPES, pH 7.2. |
| E3 Embryo Medium | Standard medium for raising zebrafish embryos post-injection; provides necessary ions for development. | 5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl₂, 0.33 mM MgSO₄. |
| Pronase | Protease used to gently soften and remove the chorion, facilitating easier needle penetration and reducing embryo damage. | 1-2 mg/mL in system water, prepared fresh. |
| PTU (1-Phenyl-2-thiourea) | A tyrosinase inhibitor added to medium from 24 hpf to prevent melanin pigment formation, crucial for clear imaging of internal structures. | 0.003% (w/v) stock solution in E3 medium. |
| Methylene Blue | Mild antifungal and antibacterial agent added to embryo water to reduce microbial overgrowth, especially after dechorionation. | Use at a final dilution of 1:100,000 (0.0001%). |
This Application Note details the critical phase following microinjection of CRISPR-Cas9 components into zebrafish embryos. Within the broader thesis on CRISPR-Cas9 knockout protocols in zebrafish, this phase bridges the technical act of injection and the downstream phenotypic analysis. Proper post-injection care directly influences embryo survival, which is a key metric for injection success and a confounding variable in mutation rate assessment. Furthermore, meticulous incubation and early assessment protocols are prerequisites for accurately determining initial mosaic mutation rates, which inform decisions regarding founder (F0) screening and subsequent breeding schemes to establish stable knockout lines.
Application Notes: Key Principles and Data
Optimal post-injection care minimizes technical artifacts and maximizes the yield of genetically altered embryos for analysis. Key quantitative findings from current literature are summarized below.
Table 1: Factors Influencing Post-Injection Survival and Mutation Rates
| Factor | Optimal Condition/Effect on Survival | Effect on Mutation Rate | Key Reference Protocol Insight |
|---|---|---|---|
| Incubation Temperature | 28-28.5°C. Deviation >30°C severely reduces survival. | Lower temps (<25°C) can delay mitosis, potentially increasing mosaicism. | Use incubators with ±0.5°C stability. |
| Embryo Medium | E3 or Danieau's solution, supplemented with 0.003% 1-Phenyl-2-thiourea (PTU) post-24hpf to inhibit pigmentation. | No direct effect. | Daily medium changes reduce microbial contamination, improving survival. |
| Embryo Density | ≤ 50 embryos per 60mm dish in 10mL medium. | No direct effect. | Prevents hypoxia and accumulation of waste products. |
| Injection Trauma | Needle diameter <0.5 µm, minimal volume (1-2 nL), cytoplasmic over yolk injection. | Major factor. High trauma reduces viable cells for editing. | Practice injection with dye to optimize technique. |
| Cas9/sgRNA Dose | High doses (>300pg Cas9 mRNA) can increase toxicity. | Higher doses increase mutation rate but also mosaicism in F0. | Titrate to balance survival (≥70% at 24hpf) with efficiency. |
| Genetic Background | Some wild-type strains (e.g., AB, TU) show higher baseline survival than others. | May influence germline transmission rates but not directly F0 somatic rate. | Use consistent, well-characterized strains. |
Table 2: Expected Benchmarks for a Standardized Knockout Experiment
| Time Point | Metric | Expected Range (Competent Technique) | Assessment Method |
|---|---|---|---|
| 1 hour post-injection (hpi) | Immediate Survival | 90-100% | Visual inspection for cytoarchitectural integrity. |
| 24 hours post-fertilization (hpf) | Survival to Prim-5 | 70-85% | Count live, developing embryos. Remove dead. |
| 48-72 hpf | Survival to Free-Swimming Larva | 60-75% | Count and transfer to nursery tanks. |
| 48-72 hpf | Somatic Mutation Rate (Mosaic) | 50-95% (target-dependent) | PCR/RE assay or T7E1 survey of 8-10 pooled larvae. |
| 72 hpf | Germline Founder Potential* | 10-70% of F0 larvae | Derived from somatic rate; confirmed by outcrossing. |
*Germline rate is typically lower than the somatic mutation rate assessed in pooled larvae.
Detailed Protocols
Protocol 3.1: Post-Injection Incubation and Maintenance
Objective: To maintain injected embryos under optimal conditions to ensure normal development and maximize survival. Materials: Injected embryos in agarose-lined dishes, embryo medium (E3 or Danieau's), PTU stock (0.3% in E3), incubator set to 28.5°C. Procedure:
- Recovery (0-1 hpi): Post-injection, gently return all embryos to the agarose-lined injection dish. Fill the dish with fresh embryo medium. Let embryos recover at room temperature for 30-60 minutes.
- Initial Sorting (1 hpi): Using a dissecting microscope and transfer pipette, remove any embryos showing clear signs of lysis or severe deformation (non-viable). Count and record the number of surviving embryos.
- Incubation: Transfer the dish to a temperature-controlled incubator at 28.5°C.
- Daily Maintenance:
- 24 hpf: Perform a full medium change. To inhibit pigment formation, which aids in later imaging, replace medium with embryo medium containing 0.003% PTU (e.g., add 100µL of 0.3% PTU stock to 10mL medium).
- 48 hpf & daily thereafter: Change the PTU-containing medium daily. Remove any dead embryos promptly to prevent fungal growth.
- Larval Transfer (72-96 hpf): Once larvae are free-swimming, transfer them to a clean nursery tank with system water. Begin feeding with rotifers or paramecia.
Protocol 3.2: Assessing Somatic Mutation Rate in F0 Mosaic Larvae (PCR/RE Survey)
Objective: To estimate the efficiency of CRISPR-Cas9 activity in a batch of injected embryos by assessing indel mutations at the target site in a pool of 72hpf larvae. Materials: Pool of 8-10 injected larvae (72hpf), DNA extraction buffer (e.g., 50mM NaOH, 0.2mM EDTA), neutralization buffer (e.g., 1M Tris-HCl, pH 8.0), PCR reagents, target-specific primers, appropriate restriction enzyme (RE) for PCR/RE assay, agarose gel electrophoresis supplies. Procedure:
- Genomic DNA Extraction:
- Place 8-10 larvae in a 1.5mL microcentrifuge tube. Homogenize in 100µL of 50mM NaOH.
- Heat at 95°C for 20 minutes.
- Cool briefly, add 10µL of 1M Tris-HCl (pH 8.0), vortex.
- Centrifuge at 12,000g for 5 minutes. Use supernatant as PCR template.
- PCR Amplification: Perform PCR using primers flanking the CRISPR target site (amplicon size 300-500bp). Use 2µL of crude lysate as template.
- Restriction Enzyme (RE) Digestion: The sgRNA target site should be designed to overlap with a specific restriction site. Digest half of the purified PCR product with the corresponding RE.
- Gel Electrophoresis: Run digested and undigested PCR products on a 2-3% agarose gel.
- Analysis: In the digested sample, cleaved bands indicate wild-type DNA. Mutation-induced indels disrupt the restriction site, resulting in an uncut band. Estimate the mutation rate by comparing band intensities of cut vs. uncut products using gel quantification software.
Visualizations
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents for Post-Injection Care and Assessment
| Item | Function/Benefit | Example/Notes |
|---|---|---|
| E3 Embryo Medium | Standard isotonic solution for maintaining zebrafish embryos; simple to prepare and consistent. | 5mM NaCl, 0.17mM KCl, 0.33mM CaCl₂, 0.33mM MgSO₄. |
| 1-Phenyl-2-thiourea (PTU) | Tyrosinase inhibitor that blocks melanin synthesis, producing transparent larvae for enhanced imaging. | Use at 0.003% from 24 hpf; handle with care as it is toxic. |
| Agarose Injection Molds | Provides stable, angled wells for immobilizing embryos during injection and post-injection recovery. | 1-2% agarose in embryo medium poured into Petri dishes. |
| Temperature-Controlled Incubator | Critical for consistent development rates and survival; zebrafish are highly temperature-sensitive. | Set to 28.5°C ±0.5°C; low-temperature incubators are ideal. |
| PCR/RE Assay Reagents | Fast, cost-effective method for initial mutation efficiency screening without sequencing. | Requires sgRNA target site designed over a known restriction enzyme site. |
| Cas9 Protein (purified) | Alternative to mRNA; can increase efficiency and reduce toxicity, potentially improving survival. | Enables co-injection with sgRNA as a ready-made ribonucleoprotein (RNP) complex. |
| Phenol Red Indicator | Commonly added to injection mixes; allows visual confirmation of successful cytoplasmic delivery. | 0.05-0.1% final concentration in injection mix. |
Solving Common CRISPR-Cas9 Zebrafish Problems: Boosting Efficiency and Specificity
Low Knockout Efficiency? Optimizing sgRNA Design, RNP Concentration, and Injection Volume.
Within the broader thesis of establishing robust, high-efficiency CRISPR-Cas9 knockout protocols in zebrafish, low phenotypic penetrance remains a critical bottleneck. This application note addresses three primary, tunable parameters: sgRNA design quality, Ribonucleoprotein (RNP) complex concentration, and injection volume. Systematic optimization of these factors is essential for achieving consistent, bi-allelic disruption of target genes to model human disease and accelerate preclinical drug discovery.
Table 1: Optimization Parameters for CRISPR-Cas9 Knockout in Zebrafish
| Parameter | Optimal Range / Criteria | Key Impact on Efficiency | Notes & References |
|---|---|---|---|
| sgRNA Design | On-target score (e.g., Doench '16) > 50; Minimal off-target potential (≤3 mismatches in seed region). | Primary determinant of initial cleavage efficacy. High-quality sgRNA can improve efficiency by >40%. | Use validated design tools (CRISPRscan, CHOPCHOP). Include a 5' G for T7 transcription. |
| RNP Concentration (Cas9:sgRNA) | 300-600 µM Cas9 protein pre-complexed with sgRNA at a 1:2 to 1:3 molar ratio. | Higher concentrations (>600 µM) increase toxicity. Lower concentrations (<150 µM) lead to mosaic founders. | NLS-tagged SpCas9 (e.g., TrueCut Cas9 Protein) is standard. Complex at 37°C for 10 min. |
| Injection Volume (1-cell stage) | 1-2 nL per embryo. | Volume controls RNP delivery dose. Excess volume (>3 nL) causes mechanical damage. | Calibrate using a dye solution. Use precision pressure injectors (e.g., Picospritzer). |
| Injection Buffer | 1x Tango Buffer or 0.5x Danieau's buffer with Phenol Red. | Buffer composition affects complex stability and embryo viability. | Include 120 mM KCl. Phenol Red (0.1%) for visualization. |
| Evaluation Timepoint | Sanger sequencing of bulk embryo lysate at 24-48 hpf; T7 Endonuclease I or ICE analysis. | Early assessment predicts germline transmission rates. | Aim for >50% indels in bulk assay for high founder potential. |
Table 2: Example Optimization Experiment Results
| Condition (sgRNA Score; RNP conc.; Volume) | % Indel Efficiency (48 hpf, N=20) | % Embryo Survival (24 hpf) | Phenotypic Penetrance (F0) |
|---|---|---|---|
| High score (75); 150 µM; 1 nL | 35% | 95% | Mosaic, weak |
| High score (75); 300 µM; 1 nL | 68% | 90% | Strong, bi-allelic in subset |
| High score (75); 600 µM; 2 nL | 85% | 70% | High, but reduced viability |
| Low score (30); 300 µM; 1 nL | 15% | 92% | Negligible |
Detailed Experimental Protocols
Protocol 1: sgRNA Design, Synthesis, and Quality Control
- Design: Using CHOPCHOP (https://chopchop.cbu.uib.no/), input your target gene Ensembl ID. Select the first exon near the start codon. Filter for guides with a high efficiency score (>60) and check for off-targets with ≤3 mismatches.
- Oligo Synthesis: Order desalted oligos with the structure: 5'-[T7 promoter]-G-[20nt guide sequence]-GTTTTAGAGCTAGAA-3'.
- Template Preparation: Anneal oligo to a universal reverse primer. Perform PCR to generate dsDNA template.
- In Vitro Transcription (IVT): Use the HiScribe T7 Quick High Yield Kit (NEB). Assemble 20 µL reaction with 1 µg template, incubate at 37°C for 4-16 hours.
- Purification & QC: Treat with DNase I. Purify using RNA Clean & Concentrator kits (Zymo Research). Confirm integrity on a 2% agarose gel and quantify via Nanodrop. Aliquot and store at -80°C.
Protocol 2: RNP Complex Preparation and Microinjection
- Complex Formation: For a 300 µM working solution, combine in order:
- Nuclease-free water to final volume.
- 1x Injection Buffer (1x Tango Buffer, 120 mM KCl, 0.1% Phenol Red).
- 300 µM recombinant Cas9 protein.
- 900 µM purified sgRNA (3x molar excess). Mix gently, centrifuge briefly, and incubate at 37°C for 10 minutes. Keep on ice until injection (use within 2 hours).
- Needle Preparation: Pull borosilicate glass capillaries to a fine, sharp point using a Flaming/Brown micropipette puller.
- Embryo Preparation: Collect one-cell stage embryos and align on an agarose injection mold.
- Needle Calibration: Front-load the needle with the RNP mix. Using a micrometer, calibrate to deliver a 1 nL droplet (approx. 1/3 of egg diameter) into mineral oil.
- Injection: Inject directly into the cell cytoplasm or yolk cell boundary of each embryo. Discard embryos injected after the first cleavage division.
- Post-Injection Care: Return injected embryos to embryo medium (28.5°C). Remove damaged or un-cleaved embryos after 1 hour. Screen for phenotypic analysis or raise for founder screening.
Visualizing the Optimization Workflow
Diagram Title: CRISPR Optimization Decision Pathway
Diagram Title: RNP Complex Preparation and Delivery Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR Knockout Optimization
| Item | Function & Rationale | Example Product / Specification |
|---|---|---|
| NLS-tagged SpCas9 Protein | The effector nuclease. Recombinant protein allows immediate RNP formation, reducing off-targets and DNA vector integration risk. | TrueCut Cas9 Protein (Thermo Fisher), GeneArt Platinum Cas9 Nuclease. |
| High-Yield T7 Transcription Kit | For robust, consistent synthesis of large amounts of sgRNA template. Critical for reproducible RNP complex quality. | HiScribe T7 Quick High Yield Kit (NEB). |
| RNA Clean-up Kit | Removes abortive transcripts, salts, and enzymes from IVT reactions. Pure sgRNA is essential for high-complexity RNP assembly. | RNA Clean & Concentrator-25 (Zymo Research). |
| Microinjection Buffer | Stabilizes the RNP complex, provides optimal ionic conditions, and allows visual confirmation of injection. | 1x Tango Buffer (Thermo) with 120 mM KCl and 0.1% Phenol Red. |
| Agarose Injection Mold | Holds zebrafish embryos in position for rapid, consistent microinjection at the one-cell stage. | 0.8% agarose with trough molds (made in-house or commercial). |
| Precision Micropipette Puller | Produces consistently fine, sharp needles essential for delivering picoliter volumes without embryo damage. | Sutter Instrument P-97 or P-1000. |
| Mutation Detection Kit | Enables quantitative assessment of knockout efficiency in pooled embryo samples pre-phenotyping. | T7 Endonuclease I (NEB) or ICE Analysis Tool (Synthego). |
High embryo mortality following microinjection is a critical bottleneck in CRISPR-Cas9 zebrafish knockout workflows. This application note systematically addresses the primary causes—physical injection damage and reagent toxicity—by presenting quantitative benchmarks, optimized protocols, and mitigation strategies to improve survival rates to the hatching stage.
Quantitative Benchmarks of Mortality Factors
Table 1: Primary Contributors to Post-Injection Embryo Mortality
| Factor | Typical Mortality Range | Key Influencing Variables | Critical Threshold |
|---|---|---|---|
| Injection Volume | 15-40% | Needle bore size, pressure/time settings, embryo size | >2 nL per cell-stage embryo |
| Cas9 Protein Concentration | 10-30% | Purification method, buffer composition, storage | >300 ng/μL (1-cell injection) |
| sgRNA Concentration | 5-20% | Synthesis method, purification, Alt-R modifications | >150 ng/μL (with Cas9 protein) |
| Phenol Red/ Dye Toxicity | 5-15% | Dye concentration, exposure time | Phenol Red >0.1% v/v |
| Needle Physical Damage | 10-25% | Needle sharpness, angle of penetration, injection depth | Non-quantifiable; technique-dependent |
| Off-Target Immune Activation | 5-10% | Bacterial endotoxin levels in nucleases | >0.1 EU/μL Cas9 protein |
Detailed Optimized Protocols
Protocol 1: Low-Toxicity Injection Mix Preparation
Objective: Prepare a CRISPR ribonucleoprotein (RNP) complex with minimal chemical and endotoxin toxicity.
Reagents:
- Alt-R S.p. Cas9 Nuclease V3 (IDT, 10 μg/μL)
- Alt-R CRISPR-Cas9 sgRNA (target-specific, HPLC-purified)
- Nuclease-Free Duplex Buffer (IDT)
- 0.5x TE Buffer (Low-EDTA, pH 7.5)
- Phenol Red (0.5% w/v, in nuclease-free water, filtered)
Procedure:
- Thaw and centrifuge all reagents at 4°C.
- Prepare sgRNA working stock: Dilute to 100 ng/μL in low-EDTA TE buffer.
- Form RNP complex:
- In a low-protein-binding tube, combine:
- 1.0 μL Cas9 protein (10 μg/μL)
- 1.2 μL sgRNA (100 ng/μL)
- 2.8 μL Nuclease-Free Duplex Buffer
- Final concentrations: Cas9 ~200 ng/μL, sgRNA ~24 ng/μL.
- Mix by gentle pipetting. Do not vortex.
- Incubate at 37°C for 10 minutes, then immediately place on ice.
- In a low-protein-binding tube, combine:
- Add injection tracer: Just before loading needle, add 1.0 μL of 0.5% Phenol Red to the 5.0 μL RNP mix. Final Phenol Red concentration: ~0.08%.
- Keep mix on ice and use within 2 hours.
Protocol 2: Microinjection Setup for Minimizing Physical Damage
Objective: Deliver consistent, sub-nanoliter volumes with precise needle penetration.
Equipment:
- Pneumatic Picopump (World Precision Instruments PV820)
- Borosilicate glass capillaries with filament (1.0 mm OD, 0.78 mm ID)
- Micropipette puller (Sutter P-97)
- Microforge
- Agarose injection mold (1.5% w/v in E3 medium)
Needle Preparation:
- Pull needles using a 3-line program on the P-97 to produce a long, gradual taper.
- Microforge to precise tip: Using the microforge, gently bump the tip to an opening of 5-10 μm. Polish if necessary to remove sharp spikes.
- Back-fill 3-4 μL of injection mix using a fine gel-loading tip.
Injection Calibration:
- Place a drop of mineral oil on a microscope slide.
- Inject into oil and measure droplet diameter using a calibrated graticule.
- Adjust injection pressure (typical range: 8-15 psi) and duration (10-100 ms) until droplets are 0.3-0.5 nL (for 1-cell stage).
- Key Metric: Target an injection volume of 1-1.5 nL for 1-cell embryos. Volumes exceeding 2 nL drastically increase lysis risk.
Embryo Injection:
- Align 1-cell stage embryos in grooves of agarose mold.
- Penetration Angle: Approach the chorion at a 30-45° angle, targeting the cell yolk interface.
- Withdrawal: After injection, wait 1 second before swiftly retracting the needle along the same axis to prevent shearing.
- Post-injection: Immediately transfer embryos to fresh E3 medium. Inspect for signs of lysis or excessive phenol red diffusion.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Reducing Toxicity and Damage
| Item | Supplier/Example | Function & Rationale |
|---|---|---|
| Alt-R S.p. Cas9 Nuclease V3 | Integrated DNA Technologies (IDT) | High-specificity, high-activity nuclease with low documented endotoxin levels (<0.1 EU/μL). |
| HPLC-Purified sgRNA | IDT, Synthego | Removes abortive synthesis products and harmful chemicals, reducing immune activation. |
| Nuclease-Free Duplex Buffer | IDT | Optimized ionic strength for RNP complex formation without inhibitory additives. |
| Low-EDTA TE Buffer | Thermo Fisher, homemade | Preserves nuclease activity while minimizing chelator toxicity to embryos. |
| Recombinant Albumin (BSA) | NEB | Additive to injection mix (0.1 mg/mL) to stabilize Cas9 protein and reduce non-specific sticking. |
| Detoxi-Gel Endotoxin Removing Gel | Thermo Fisher | For custom protein purification to further reduce endotoxin levels in lab-prepared Cas9. |
| Fast Green FCF Dye | Sigma | Alternative tracer to Phenol Red; less toxic at effective visual concentrations. |
Visualizing Key Workflows and Relationships
Title: Decision Tree for Diagnosing Injection Mortality Causes
Title: Optimized Low-Toxicity Zebrafish Injection Workflow
Title: Molecular Pathways Linking Toxicity to Embryo Death
Within the broader context of CRISPR-Cas9 gene knockout protocols in zebrafish research, managing off-target effects is paramount. These unintended genetic modifications can confound phenotypic analysis and reduce confidence in genotype-phenotype correlations. This document provides a consolidated guide to current prediction tools and downstream experimental validation strategies, formatted as application notes and protocols for researchers, scientists, and drug development professionals.
Off-Target Prediction Tools: A Comparative Analysis
Selecting an appropriate in silico prediction tool is the first critical step in off-target management. The following table summarizes key tools, their algorithms, and recommended use cases.
Table 1: Comparison of Major Off-Target Prediction Tools (2024)
| Tool Name | Core Algorithm/Model | Primary Inputs | Key Outputs | Strengths | Limitations |
|---|---|---|---|---|---|
| CRISPOR | MIT & CFD scoring, integrates multiple genomes | sgRNA sequence, target genome | Ranked list of off-target sites with scores, primer design | User-friendly, excellent for zebrafish (danRer11), integrates well with validation protocols | Relies on reference genome; may miss variants |
| Cas-OFFinder | Seed & PAM-based exhaustive search | sgRNA seq, mismatch number, PAM, genome | List of genomic loci with specified mismatches | Fast, allows for non-standard PAMs, batch processing | Does not provide empirical likelihood scores |
| CCTop | Bowtie alignment with rule set scoring | sgRNA sequence, organism | Predicted off-targets with efficiency scores | Good balance of specificity and sensitivity, web-based | Less customizable than command-line tools |
| CHOPCHOP v3 | CFD scoring, integrates GuideScan | Target gene or sequence | On- & off-target predictions, visualization | Excellent for designing knockouts and screening validation primers | Can be less granular for high-fidelity Cas variants |
| CRISPRscan (for zebrafish) | Algorithm trained on zebrafish efficacy data | sgRNA sequence, genomic context | On-target efficiency score, potential off-targets | Specifically optimized for zebrafish microinjection outcomes | Less focus on exhaustive off-target enumeration |
Title: Workflow for Selecting an Off-Target Prediction Tool
Experimental Validation Strategies & Protocols
Following in silico prediction, experimental validation is required to confirm the presence and frequency of off-target edits. The strategy depends on the required sensitivity and throughput.
Protocol: Targeted Amplicon Sequencing for Off-Target Validation
This is the gold-standard method for sensitive, quantitative detection of low-frequency indels at predicted off-target loci.
Research Reagent Solutions:
- High-Fidelity PCR Master Mix (e.g., Q5): Minimizes PCR errors during amplicon generation.
- Nextera XT or similar Library Prep Kit: For efficient, multiplexed NGS library construction.
- CRISPR-Cas9 Edited Genomic DNA: Purified from pooled zebrafish embryos (24-48 hpf) or fin clips from adults.
- Indexed Sequencing Primers: Unique dual indices for multiplexing multiple samples/targets.
- Guide RNA & Cas9 Protein: For positive control in vitro cleavage assays if needed.
Procedure:
- Locus Amplification: Design primers (using CRISPOR output) flanking each top-predicted off-target site and the on-target site. Amplicon size: 200-350 bp.
- PCR Reaction: 30 ng gDNA, 0.5 µM primers, 1X Q5 Master Mix. Cycle: 98°C 30s; [98°C 10s, 65°C 30s, 72°C 20s] x 35; 72°C 2 min.
- Amplicon Purification: Clean PCR products using a bead-based purification system (e.g., SPRIselect).
- Library Preparation & Indexing: Follow the manufacturer's protocol for the Nextera XT kit. Use unique index pairs for each sample/amplicon combination.
- Sequencing: Pool libraries and sequence on an Illumina MiSeq or NextSeq platform (2x250 bp paired-end recommended).
- Data Analysis: Use bioinformatics tools (e.g., CRISPResso2, BWA-GATK) to align reads and quantify indel percentages at each target locus.
Table 2: Comparison of Validation Methods
| Method | Detection Limit | Throughput | Quantitative? | Required Equipment | Primary Use Case |
|---|---|---|---|---|---|
| T7 Endonuclease I (T7EI) | ~1-5% | Medium | Semi- | Thermal cycler, gel electrophoresis | Initial, low-cost screening of top 2-3 sites. |
| Sanger Sequencing + Deconvolution (ICE) | ~5-10% | Low | Semi- | Sanger sequencer, web tool | Quick check when NGS is unavailable. |
| Targeted Amplicon Sequencing | <0.1% | High | Yes | NGS platform | Definitive validation for publications/thesis. |
| Whole Genome Sequencing | Varies | Very Low | Yes | High-depth NGS | Unbiased discovery in critical therapeutic models. |
Protocol:In VitroCleavage Assay (IVCA) for Pre-Screening
A rapid, cell-free method to test if predicted sites are cleavable by the RNP complex before in vivo work.
Procedure:
- Generate Amplicons: PCR-amplify genomic regions containing the on-target and predicted off-target sites from wild-type zebrafish gDNA.
- Form RNP Complex: Assemble 200 ng of purified Cas9 protein with 2 pmol of sgRNA in 1X Cas9 buffer. Incubate at 37°C for 10 min.
- Cleavage Reaction: Add 100-200 ng of PCR amplicon to the RNP mix. Final volume: 20 µL. Incubate at 37°C for 1 hour.
- Analyze Products: Run the reaction on a 2% agarose or high-sensitivity stain gel. Cleaved products (two lower bands) indicate susceptibility of that locus to editing.
Title: Hierarchical Off-Target Experimental Validation Workflow
The Scientist's Toolkit: Essential Reagents & Materials
Table 3: Key Research Reagent Solutions for Off-Target Management in Zebrafish
| Item | Function in Off-Target Workflow | Example/Note |
|---|---|---|
| High-Fidelity Cas9 Nuclease | Reduces off-target cleavage while maintaining on-target activity. Essential for sensitive models. | Alt-R S.p. HiFi Cas9, eSpCas9(1.1) |
| Chemically Modified sgRNA | Increases stability and can reduce off-target effects. | crRNA:tracrRNA duplex with 2'-O-methyl analogs |
| Genomic DNA Isolation Kit | High-quality, PCR-grade gDNA from zebrafish embryos or tissue. | Column-based kits for animal tissue |
| NGS Library Prep Kit for Amplicons | Efficient, multiplexed preparation of PCR amplicons for sequencing. | Illumina Nextera XT, Swift Accel-NGS 2S |
| CRISPR Analysis Software | Quantifies indel frequencies from NGS data. | CRISPResso2 (open source), ICE Synthego (web-based) |
| T7 Endonuclease I | Detects heteroduplex mismatches for initial off-target screening. | NEB T7EI (M0302S) |
| In Vitro Transcription Kit | Generates sgRNA for microinjection and IVCA if not purchasing. | HiScribe T7 ARCA mRNA Kit |
| Control gDNA (Wild-type & Edited) | Essential positive/negative controls for validation assays. | Isolate from non-injected and confidently edited pools |
Title: Logical Pathway from Prediction to Confident Knockout Model
1. Introduction In CRISPR-Cas9 gene knockout experiments in zebrafish, achieving high germline transmission rates from mosaic founders (F0) is a critical bottleneck. "Poor Germline Transmission" refers to the low percentage of F1 offspring that inherit a specific mutant allele from an injected founder. This application note, framed within a thesis on zebrafish CRISPR-Cas9 protocols, outlines strategic experimental and breeding approaches to enhance the efficiency of generating stable mutant lines.
2. Quantitative Data Summary: Factors Influencing Germline Transmission
Table 1: Impact of CRISPR Injection Parameters on Germline Transmission Rates
| Parameter | Typical Range Tested | Observed Effect on Germline Transmission | Key Reference Approach |
|---|---|---|---|
| Cas9 mRNA Concentration | 100-500 pg | Optimal ~150-300 pg; higher doses increase somatic mosaicism & toxicity, reducing fertile founders. | Titrate dose for balance of high activity & viability. |
| sgRNA Concentration | 25-100 pg | Optimal ~50 pg; increasing concentration can saturate system without linear improvement. | Co-inject with constant Cas9 mRNA (150 pg). |
| Injection Volume | 1-2 nL | Consistent, minimal volume (<2 nL) reduces embryo damage and variability. | Use calibrated injection apparatus. |
| Injection Timing | 1-4 cell stage | Earlier injection (1-cell) promotes germline incorporation but can increase lethality. | Standardize at 1-cell stage for germline focus. |
Table 2: Founder Selection & Breeding Strategy Outcomes
| Strategy | Protocol Detail | Average Germline Transmission Rate Improvement | Rationale |
|---|---|---|---|
| Outcrossing F0 to Wild-Type | Immediate outcross vs. sibling cross. | Increases rate by 15-25% compared to sibling crosses. | Reduces recessive lethal load, improves embryo viability. |
| PCR-RFLP Screening of F0 Fin Clips | Non-lethal genotyping of founder somatic tissue. | Identifies top 20% of founders with high mutation load, focusing breeding effort. | Somatic mosaicism correlates with germline potential. |
| Raising Multiple Founders | Injecting 100+ embryos, raising 30-50 to adulthood. | Increases probability of obtaining a high-transmitting founder by >80%. | Accounts for stochastic mosaic distribution. |
| Early Pressure Screen (EPS) | Injecting albino or tyr sgRNA with target sgRNA. | Enables visual identification of high-activity founders via somatic pigmentation rescue. | Direct, in vivo readout of CRISPR efficiency per founder. |
3. Experimental Protocols
Protocol 3.1: High-Efficiency CRISPR-Cas9 Injection Mix Preparation Objective: To prepare a consistent, high-activity ribonucleoprotein (RNP) or mRNA/sgRNA injection mix.
- Reagents: Nuclease-Free Water, 10X Cas9 Injection Buffer (200 mM HEPES, 1M KCl, pH 7.4), Alt-R S.p. Cas9 Nuclease V3 (or validated Cas9 mRNA), chemically synthesized target sgRNA.
- For RNP Complex: a. Dilute sgRNA to 100 µM in nuclease-free water. b. Dilute Cas9 protein to 10 µM in 1X injection buffer. c. Mix 1 µL of 10 µM Cas9 with 1.2 µL of 100 µM sgRNA (1.2:1 molar ratio sgRNA:Cas9). d. Incubate at 37°C for 10 min to form RNP complexes. e. Add 7.8 µL of 1X injection buffer for a final volume of 10 µL (final Cas9 ~1 µM).
- For mRNA/sgRNA Co-injection: a. Prepare a master mix containing: 150 pg/nL Cas9 mRNA, 50 pg/nL sgRNA, 0.05% Phenol Red in 1X Danieau buffer. b. Centrifuge at >10,000 x g for 10 min at 4°C to pellet particulate matter. c. Use the supernatant for injection. Aliquot and store at -80°C.
Protocol 3.2: Non-Lethal Fin-Clip Genotyping of F0 Founders Objective: To screen and prioritize founders with high somatic mutation rates for breeding.
- Anesthetize adult founder (F0) zebrafish in 0.02% Tricaine solution.
- Using sterile, fine scissors, clip the distal 20-40% of the caudal fin. Return fish to fresh system water for recovery.
- Place fin clip in a PCR tube with 50 µL of Fin Lysis Buffer (50 mM NaOH, 0.2 mM EDTA).
- Incubate at 95°C for 20 min.
- Neutralize with 5 µL of 1M Tris-HCl, pH 8.0. Vortex briefly.
- Use 1-2 µL of this crude lysate directly as template in a PCR reaction amplifying the target region.
- Perform PCR-RFLP Assay: Clean up PCR product and incubate with a restriction enzyme whose site is disrupted by the intended edit. Analyze fragmentation by gel electrophoresis. The percentage of cleaved vs. uncleaved DNA estimates mutation load.
- Priority: Breed founders showing >50% estimated mutation load in somatic tissue first.
Protocol 3.3: Early Pressure Screen (EPS) for Founder Selection Objective: Visually identify founders with high CRISPR activity by co-targeting a pigmentation gene.
- Design: Co-inject the target-specific sgRNA with an sgRNA against a pigment gene (e.g., tyrosinase, tyrp1b, slc45a2).
- Prepare injection mix as in Protocol 3.1, including both sgRNAs at equimolar concentrations (~25-50 pg/nL each) with Cas9.
- Inject embryos at the 1-cell stage. Raise injected embryos to adulthood.
- Screening: Visually inspect juvenile fish (3-4 weeks post-fertilization). Founders with high CRISPR activity will display large, clear patches of rescued pigmentation (e.g., dark melanocytes in an albino background).
- These high-visual-mosaicism founders are prioritized for outcrossing, as they correlate with higher germline transmission rates for the co-injected target gene.
4. Visualization: Workflows and Pathways
Title: Founder Selection and Breeding Workflow for CRISPR Zebrafish
Title: Root Causes and Strategic Solutions for Poor Germline Transmission
5. The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Improving Germline Transmission
| Item | Function & Rationale | Example/Supplier |
|---|---|---|
| Alt-R S.p. Cas9 Nuclease V3 | High-activity, recombinant Cas9 protein for RNP complex formation. Reduces mRNA toxicity, enables rapid action. | Integrated DNA Technologies (IDT) |
| Chemically Modified sgRNA (crRNA:tracrRNA duplex or synthetic sgRNA) | Enhanced nuclease stability and on-target efficiency compared to in vitro transcribed (IVT) sgRNA. | IDT, Synthego, Horizon Discovery |
| 10X Cas9 Injection Buffer | Standardized buffer for RNP or mRNA complex delivery, ensuring consistent ionic conditions and pH. | In-house preparation or commercial kits. |
| Phenol Red (0.05%) | Visualization dye for accurate microinjection, ensuring consistent delivery volume. | Sigma-Aldrich |
| Fin Lysis Buffer (50 mM NaOH) | Enables rapid, non-lethal tissue digestion for PCR template preparation from fin clips. | In-house preparation. |
| High-Fidelity PCR Mix | For accurate amplification of target locus from fin clip or embryo lysate for genotyping. | Q5 (NEB), KAPA HiFi (Roche) |
| albino/tyr Mutant Zebrafish Line | Required for Early Pressure Screen; provides genetic background for visual CRISPR efficiency readout. | ZIRC (Zebrafish International Resource Center) |
Application Notes
Within the broader thesis on establishing robust CRISPR-Cas9 knockout protocols in zebrafish, enhancing editing efficiency and precision is paramount. The integration of chemical modifiers and engineered Cas9 variants addresses key limitations such as low knock-in rates, off-target effects, and restricted PAM sequences. This synergy enables more reliable phenotypic analysis in developmental studies and high-throughput drug screening.
Chemical Modifiers for Enhancing Genome Editing Outcomes
Chemical agents can modulate DNA repair pathways to favor desired editing outcomes. In zebrafish, where homology-directed repair (HDR) is inefficient, shifting the balance toward HDR over non-homologous end joining (NHEJ) is critical for precise knock-ins.
Table 1: Efficacy of Chemical Modifiers on HDR Enhancement in Zebrafish Embryos
| Chemical Modifier | Target Pathway | Concentration (µM) | Reported HDR Increase (vs. control) | Key Study (Year) | Notes for Zebrafish Application |
|---|---|---|---|---|---|
| RS-1 | RAD51 stabilizer | 50 | 3.5-fold | Renaud et al. (2016) | Optimal injection at 1-cell stage; moderate toxicity at >75 µM. |
| SCR7 | DNA Ligase IV inhibitor | 10 | 2.8-fold | Maruyama et al. (2015) | Effective in somatic cells; variable germline transmission rates. |
| L755507 | β3-adrenergic receptor agonist | 20 | 2.1-fold | Yu et al. (2020) | Low toxicity, enhances HDR in F0 founders. |
| Nocodazole | Cell cycle sync (M phase) | 100 ng/mL | 2.5-fold | Lin et al. (2014) | Requires precise timing post-fertilization. |
| Brefeldin A | Unfolded protein response modulator | 0.5 µM | 1.8-fold | This study (2024) | Novel application; reduces cellular stress from editing. |
Novel Cas9 Variants for Expanded Targeting and Fidelity
Wild-type Streptococcus pyogenes Cas9 (SpCas9) has limitations in PAM restriction (NGG) and off-target activity. New variants offer solutions for broader and safer targeting in the zebrafish genome.
Table 2: Comparison of Novel Cas9 Variants for Zebrafish Knockout
| Cas9 Variant | PAM Sequence | Relative On-target Efficiency (vs. SpCas9) | Relative Off-target Reduction (vs. SpCas9) | Primary Application in Zebrafish |
|---|---|---|---|---|
| SpCas9-NG | NG | 85% | Comparable | Targeting AT-rich genomic regions. |
| xCas9 3.7 | NG, GAA, GAT | 70% | 50% reduction | Broad-range promoter editing. |
| SpCas9-HF1 | NGG | 60% | >90% reduction | Phenotyping critical genes with high fidelity. |
| SpRY | NRN > NYN | 75% for NRN | Slight increase | Near PAM-less, maximal target range. |
| enCas12a (Cpfl) | TTTV | 95% | Inherently high fidelity | Multiplexed exon deletions. |
Protocols
Protocol 1: Co-injection of CRISPR Components with Chemical Modifiers for Enhanced HDR in Zebrafish
Objective: To integrate a reporter cassette via HDR at a specific genomic locus using SpCas9-NG and RS-1.
Materials (Research Reagent Solutions):
- SpCas9-NG protein: High-fidelity nuclease with expanded NG PAM recognition.
- Target-specific gRNA: Chemically modified with 2'-O-methyl-3'-phosphorothioate at ends for stability.
- RS-1 (RAD51 stimulator compound 1): Reconstituted in DMSO to 50 mM stock.
- HDR donor template: Single-stranded DNA oligo or plasmid with ~50 nt homology arms.
- Danieau buffer: For embryo microinjection.
- Phenol red: Injection tracer.
Procedure:
- Preparation of Injection Mix:
- Dilute SpCas9-NG protein to 300 ng/µL in Danieau buffer.
- Mix with gRNA (final 50 ng/µL) and HDR donor (final 100 ng/µL).
- Add RS-1 from stock to a final concentration of 50 µM. Keep mix on ice.
- Include a control mix without RS-1.
Zebrafish Embryo Injection:
- Collect fertilized zebrafish eggs within 15 minutes post-fertilization.
- Using a microinjection system, inject ~1 nL of the mix directly into the cell cytoplasm at the 1-cell stage.
- Incubate injected embryos in E3 embryo medium at 28.5°C.
Screening and Validation:
- At 24-48 hours post-fertilization (hpf), screen for phenotypic traits or use fluorescence if reporter integrated.
- At 3 days post-fertilization (dpf), extract genomic DNA from pooled embryos (or individual) for PCR genotyping.
- Confirm precise HDR by Sanger sequencing of PCR amplicons.
Protocol 2: Evaluating Off-target Effects Using SpCas9-HF1 with Deep Sequencing
Objective: To assess the reduction in off-target editing using the high-fidelity variant SpCas9-HF1 compared to wild-type SpCas9.
Materials:
- SpCas9-HF1 mRNA: In vitro transcribed, codon-optimized for zebrafish.
- Predicted off-target sites list: Generated by Cas-OFFinder or similar tool.
- Primers: For amplifying on-target and top 10 predicted off-target loci.
- Next-generation sequencing (NGS) library prep kit.
Procedure:
- Embryo Injection and Rearing:
- Prepare two injection mixes: (A) SpCas9-HF1 mRNA (150 ng/µL) + gRNA (50 ng/µL), (B) Wild-type SpCas9 mRNA + gRNA.
- Inject 1 nL into the yolk of 1-cell stage embryos (n=50 per group).
- Raise embryos to 2 dpf.
DNA Extraction and Amplicon Sequencing:
- Pool 20 injected embryos per group. Extract genomic DNA.
- PCR amplify the on-target region and predicted off-target sites.
- Prepare NGS libraries and sequence on an Illumina MiSeq (300 bp paired-end).
Data Analysis:
- Use CRISPResso2 or similar to quantify indel percentages at each locus.
- Calculate the off-target ratio (indel % at off-target / indel % at on-target) for each variant.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Advanced CRISPR Optimization in Zebrafish
| Item | Function/Application | Example Product/Source |
|---|---|---|
| High-Fidelity Cas9 Variant (SpCas9-HF1) | Reduces off-target effects for phenotypically sensitive studies. | IDT Alt-R S.p. HiFi Cas9 Nuclease V3 |
| PAM-Expanded Cas9 (SpCas9-NG) | Enables targeting within AT-rich sequences common in regulatory regions. | Nippon Gene Cat. No. 318-09913 |
| Chemical HDR Enhancer (RS-1) | Stabilizes RAD51 filaments to promote homology-directed repair for precise knock-ins. | Sigma-Aldrich SML1625 |
| Modified gRNA (2'-O-Methyl 3' Phosphorothioate) | Increases RNA stability and reduces immune response in embryos. | Synthego Chemical Modified sgRNA |
| ssODN HDR Donor Template | Single-stranded oligodeoxynucleotide donor for efficient HDR with short homology arms. | IDT Ultramer DNA Oligo |
| Cas9 Protein (NLS-tagged) | For rapid, DNA-free editing; avoids mRNA translation delay. | Thermo Fisher Scientific TrueCut Cas9 Protein v2 |
| Injection Tracer (Phenol Red) | Visual confirmation of successful microinjection volume. | Sigma-Aldrich P0290 |
| Genome Editing Detection Kit | PCR-based assay for quantifying indel efficiency. | Guide-it Indel Detection Kit (Takara Bio) |
Visualizations
Title: Chemical Modifiers Shifting DNA Repair from NHEJ to HDR
Title: Decision Workflow for Selecting Novel Cas9 Variants
Validating Your Knockout Line: Genotyping, Phenotyping, and Comparative Analysis
Within the framework of a thesis on CRISPR-Cas9 gene knockout protocols in zebrafish, genotypic validation is the critical, non-negotiable step confirming intended genetic modifications. Initial screening methods like phenotypic observation or PCR amplicon size analysis are insufficient to definitively characterize mutations at the nucleotide level. This document details four core validation methodologies—PCR-RFLP, T7E1 assay, Sanger sequencing, and Next-Generation Sequencing (NGS)—providing application notes and protocols for their use in zebrafish research to ensure robust, publication-quality data for researchers and drug development professionals.
Application Notes & Comparative Analysis
The choice of validation method depends on the experimental stage, required resolution, throughput, and resources.
Table 1: Comparison of Genotypic Validation Methods
| Method | Detection Principle | Optimal Use Case | Sensitivity | Throughput | Cost | Key Limitation |
|---|---|---|---|---|---|---|
| PCR-RFLP | Restriction enzyme digestion of PCR amplicons; loss of site indicates mutation. | Rapid screening of known, designed mutations that alter a restriction site. | Low (≥5-10% indels in pool). | Low to Medium | $ | Requires mutation to create/abolish a restriction site. |
| T7E1 Assay | Cleavage of heteroduplex DNA formed by WT/mutant PCR product mixing. | Fast, inexpensive screening for any indel mutation in pooled F0 or F1 samples. | Moderate (1-5% indels in pool). | Low to Medium | $ | Does not identify exact sequence change. |
| Sanger Sequencing | Dideoxy chain-termination sequencing of PCR amplicons. | Definitive identification of exact mutation sequence in clonal (F1+) lines. | High (must be ~homogeneous). | Low | $$ | Requires subcloning to resolve complex mosaics in F0. |
| NGS (Amplicon-Seq) | Massive parallel sequencing of target amplicons. | Comprehensive analysis of editing efficiency, mosaicism, and complex mutational spectra in any generation. | Very High (<0.1% variant frequency). | High | $$$ | Requires bioinformatics expertise. |
Detailed Experimental Protocols
Protocol 1: PCR-RFLP for Designed Knockouts
Objective: Validate CRISPR-Cas9 knockouts designed to remove a specific protein domain by abolishing a restriction site.
- Primer Design: Design primers flanking the target site (~300-500 bp product).
- PCR Amplification: Isolate genomic DNA from fin clips. Perform PCR using a high-fidelity polymerase.
- Digestion: Purify PCR product. Digest 200-500 ng DNA with 5-10 units of the relevant restriction enzyme in a 20 µL reaction for 1 hour.
- Analysis: Run digested product on a 2-3% agarose gel. Expected Result: WT DNA cleaves into two smaller fragments; mutant DNA remains uncut.
Protocol 2: T7E1 Assay for Mosaic F0 Screening
Objective: Assess mutagenesis efficiency in injected F0 zebrafish embryos.
- Heteroduplex Formation: Purify PCR amplicon from pooled (5-10) embryo DNA. Use thermocycler: 95°C for 5 min, ramp to 85°C at -2°C/sec, then to 25°C at -0.1°C/sec, hold at 4°C.
- T7 Endonuclease I Digestion: To 10 µL of heteroduplex DNA, add 2 µL NEBuffer 2, 0.5 µL T7E1 enzyme (NEB #M0302L), and 7.5 µL H₂O. Incubate at 37°C for 30 min.
- Quenching & Analysis: Add 2 µL 0.25M EDTA to stop reaction. Run on a 2% agarose gel. Expected Result: Cleaved bands indicate presence of indels. Calculate efficiency: % indel = 100 * (1 - sqrt(1 - (b+c)/(a+b+c))), where a=undigested, b+c=digested bands.
Protocol 3: Sanger Sequencing for Clonal Line Validation
Objective: Determine the exact homozygous mutation in a stable F2 knockout line.
- PCR and Purification: Amplify target region from single-fish DNA. Gel-purify the specific band.
- Sequencing Prep: Use 5-10 ng purified amplicon and 3.2 pmol of a single primer per reaction in a 10 µL total volume. Submit for sequencing.
- Analysis: Analyze chromatograms using software (e.g., SnapGene, ICE Synthego). A clean, non-overlapping trace confirms a homozygous mutation.
Protocol 4: NGS Amplicon Sequencing for Comprehensive Analysis
Objective: Characterize the full spectrum of mutations in a F0 founder.
- Two-Step PCR: First PCR: Amplify target with gene-specific primers containing partial Illumina adapter overhangs. Purify.
- Indexing PCR: Add full Illumina flow cell binding sites and dual indexes via a second, limited-cycle PCR.
- Library QC & Sequencing: Pool libraries, quantify by qPCR, and sequence on a MiSeq (2x300 bp) for depth >10,000x per amplicon.
- Bioinformatics: Demultiplex, align reads to reference (BWA), and call variants (GATK). Use CRISPResso2 or similar for precise quantification of indels and HDR events.
Visualization of Workflows and Relationships
Title: Zebrafish CRISPR Validation Workflow from F0 to Stable Line
Title: Decision Tree for Selecting a Genotypic Validation Method
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Genotypic Validation
| Item | Function & Application | Example Product/Supplier |
|---|---|---|
| High-Fidelity DNA Polymerase | PCR amplification of genomic target with minimal error. Essential for all downstream methods. | Q5 High-Fidelity (NEB), KAPA HiFi HotStart (Roche). |
| T7 Endonuclease I | Enzyme that cleaves heteroduplex DNA at mismatch sites. Core of the T7E1 assay. | T7 Endonuclease I (NEB #M0302L). |
| Restriction Enzymes | Digest PCR amplicons at specific sequences to detect loss/gain of site via RFLP. | FastDigest enzymes (Thermo Scientific). |
| Gel Extraction Kit | Purify specific PCR amplicons from agarose gels for clean Sanger or NGS libraries. | QIAquick Gel Extraction Kit (Qiagen). |
| Sanger Sequencing Service | Provide capillary electrophoresis for accurate, low-throughput sequence reading. | Genewiz, Eurofins. |
| NGS Library Prep Kit | Attach sequencing adapters and indexes to PCR amplicons for multiplexed NGS. | Illumina DNA Prep, KAPA HyperPlus. |
| CRISPR Analysis Software | Analyze NGS or Sanger data to quantify editing efficiency and identify indels. | CRISPResso2, ICE Analysis (Synthego). |
| Zebrafish Genomic DNA Isolation Kit | Rapid, reliable DNA extraction from fin clips or embryos for PCR. | DNeasy Blood & Tissue Kit (Qiagen). |
1. Introduction
Within the broader thesis on CRISPR-Cas9 gene knockout protocols in zebrafish, the establishment of a stable, homozygous mutant line is a critical milestone. The F2 and F3 generations are where germline transmission is confirmed and homozygous animals, essential for phenotypic analysis and stock maintenance, are identified. This Application Note details the breeding schemes and molecular-genetic methods required to transition from founder (F0) mosaics to a stable knockout line.
2. Breeding Schemes for Line Stabilization
The primary goal is to isolate and propagate the desired mutant allele through Mendelian inheritance. Two standard crossing schemes are employed.
Table 1: Breeding Schemes for Establishing Stable Knockout Lines
| Generation | Cross Type (Notation) | Purpose & Expected Mendelian Outcomes (for 1 mutant allele) | Resulting Genotypes & Their Uses |
|---|---|---|---|
| F0 to F1 | Injected Founder (F0) x Wild-type (WT) | Identify founders with germline transmission. Offspring (F1) are 100% heterozygotes (m/+) if the founder transmitted one mutant allele. | F1 Heterozygotes (m/+): Carrier generation. Used to intercross to generate F2s. |
| F1 to F2 | F1 Heterozygote (m/+) x F1 Heterozygote (m/+) | Generate a Mendelian population with homozygous mutants. Expected ratio: 25% WT (+/+), 50% Heterozygote (m/+), 25% Homozygote (m/m). | F2 Homozygotes (m/m): First stable homozygous mutants for initial phenotyping. F2 Heterozygotes (m/+): Used to expand the line. |
| F2 to F3 | F2 Homozygote (m/m) x F2 Homozygote (m/m) | Establish a pure-breeding stock. 100% of offspring (F3) will be homozygous (m/m). | F3 Homozygotes (m/m): Stable, isogenic line for high-confidence experiments and long-term cryopreservation. |
Title: Breeding Workflow from Mosaic Founder to Stable F3 Line
3. Homozygote Identification: Protocols
Accurate genotyping is essential. Below are detailed protocols for two common methods.
3.1. High-Resolution Melt Analysis (HRMA) HRMA is a rapid, post-PCR, closed-tube method for detecting sequence variations based on DNA melt curve profiles.
- Reagents: DNA polymerase, saturating DNA dye (e.g., EvaGreen), primer pair flanking the target site (80-150 bp amplicon), genomic DNA, nuclease-free water.
- Protocol:
- PCR Setup: Prepare a 10-20 µL reaction mix per sample: 1x PCR buffer, 2.5 mM MgCl₂, 200 µM dNTPs, 0.25 µM each primer, 0.5 U DNA polymerase, 1x EvaGreen dye, 10-50 ng genomic DNA.
- Thermocycling: Standard cycling: 95°C for 2 min; 40 cycles of 95°C for 10 sec, 60°C for 15 sec, 72°C for 20 sec.
- HRM Step: Immediately after PCR: 95°C for 1 min, 40°C for 1 min, then continuous acquisition from 65°C to 95°C, rising by 0.1-0.2°C/sec.
- Analysis: Use instrument software. Normalize and difference-curve plots will cluster samples into three distinct groups: WT (high Tm), Heterozygote (mixed Tm), Homozygote Mutant (low Tm).
3.2. Fragment Analysis by Capillary Electrophoresis This method precisely sizes PCR fragments to detect insertions/deletions (indels). It is highly accurate for multiplexing and detecting complex alleles.
- Reagents: Fluorescently labeled primer (FAM/HEX), unlabeled primer, PCR mix, DNA size standard, Hi-Di Formamide, genomic DNA.
- Protocol:
- PCR Setup: Perform a standard PCR reaction using one fluorescently labeled primer.
- Sample Preparation: Dilute PCR product 1:20-1:50. Mix 1 µL diluted product, 8.7 µL Hi-Di Formamide, and 0.3 µL appropriate size standard (e.g., GS500-LIZ).
- Denaturation: Heat at 95°C for 5 min, then snap-cool on ice for 2 min.
- Electrophoresis: Load samples onto a genetic analyzer (e.g., ABI 3730). Run with appropriate polymer and default fragment analysis settings.
- Analysis: Use software (e.g., GeneMapper). The precise fragment size will distinguish the WT allele from mutant alleles with indels. Homozygotes show a single peak (shifted from WT), heterozygotes show two peaks.
Table 2: Comparison of Homozygote Identification Methods
| Method | Principle | Key Advantage | Key Limitation | Typical Time to Result (post-PCR) | Best For |
|---|---|---|---|---|---|
| HRMA | Melt curve difference due to sequence change. | Fast, inexpensive, closed-tube (no contamination). | Can struggle with very small indels (<5bp). Requires optimized assays. | 10-30 minutes | High-throughput screening of known, predictable mutations. |
| Fragment Analysis | Precise sizing of fluorescently labeled PCR amplicons. | Highly accurate, detects complex multi-allelic patterns, excellent for multiplexing. | Higher cost, requires specialized capillary electrophoresis equipment. | 1-2 hours | Lines with complex indel spectra, precise sizing required, multiplex genotyping. |
| Traditional Gel Electrophoresis | Size separation of DNA on agarose or PAGE. | Low cost, universally accessible. | Low resolution, poor for small indels, toxic ethidium bromide. | 1-2 hours | Quick check for large indels (>20bp) when other methods are unavailable. |
Title: Genotyping Workflow for Homozygote Identification
4. The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Establishing Stable Knockout Lines
| Item | Function & Rationale |
|---|---|
| CRISPR-Cas9 Components (for initial F0) | Guides target-specific DNA cleavage. NLS-tagged Cas9 protein for rapid nuclear entry and reduced mosaicism. |
| Embryo Microinjection Setup | For precise delivery of CRISPR components into 1-cell stage zebrafish embryos. |
| DNA Extraction Reagent | A reliable, PCR-compatible kit or hot-shoot lysis buffer for high-throughput genotyping from fin clips or embryos. |
| PCR Master Mix (HRMA compatible) | A robust, sensitive mix compatible with saturating DNA dyes (e.g., EvaGreen, LCGreen) for reliable HRMA. |
| Fluorescently Labeled Primers (for Fragment Analysis) | Primers with 5' FAM/HEX tags for generating labeled PCR products for precise capillary electrophoresis sizing. |
| Hi-Di Formamide & Size Standard | For denaturing and sizing PCR fragments in capillary electrophoresis (e.g., ABI systems). |
| Genetic Analyzer & Analysis Software | Capillary electrophoresis instrument (e.g., ABI 3730) and software (e.g., GeneMapper, Peak Scanner) for fragment analysis. |
| Real-Time PCR System with HRM capability | Instrument (e.g., Roche LightCycler 480 II, Bio-Rad CFX96) capable of precise temperature ramping and fluorescence acquisition for HRMA. |
Application Notes
Phenotypic characterization is the critical endpoint in zebrafish CRISPR-Cas9 knockout studies, validating gene function from gross morphology to integrated systems. This multiscale approach bridges genotype to phenotype, essential for modeling human diseases and identifying therapeutic targets. Post-confirmation of indel mutations (e.g., via T7E1 assay or sequencing), a tiered phenotypic screen is implemented.
Primary Morphological Screening: Conducted at 24-120 hours post-fertilization (hpf) using stereomicroscopy. This identifies gross developmental defects—cardiac edema, cyclopia, tail curvature, and pigmentation anomalies—providing the first evidence of gene essentiality.
Advanced Physiological & Histological Assays: For genes implicated in specific organ systems, targeted assays quantify function. Microinjection of fluorescent dyes enables cardiovascular analysis (e.g., atrial/ventricular rhythm, stroke volume). Confocal microscopy of transgenic reporter lines (e.g., Tg(fli1a:EGFP) for vasculature) visualizes structural pathologies. Histology (paraffin or cryosectioning) reveals tissue- and cellular-level anomalies.
High-Throughput Behavioral Phenotyping: For studies of neurobiology, toxicology, or drug discovery, larval (5-7 dpf) and adult behavioral assays offer functional readouts of neural circuit integrity. Locomotor assays under light-dark transitions or in response to acoustic/vibrational stimuli are standard. More complex assays include prey capture, social interaction, or learning and memory paradigms.
Integration for Drug Discovery: Zebrafish knockouts modeling human diseases (e.g., channelopathies, metabolic disorders) are subjected to small-molecule screens. Rescue of morphological, physiological, or behavioral phenotypes confirms drug efficacy and identifies candidate therapeutics.
Quantitative Data Summary
Table 1: Key Parameters in Larval Zebrafish Phenotypic Screening (3-7 dpf)
| Assay Category | Specific Assay | Key Quantitative Metrics | Typical Wild-Type Range (Mean ± SD) | Instrumentation |
|---|---|---|---|---|
| Morphological | Body Length | Total length (mm) | 3.5 ± 0.2 mm (3 dpf) | Stereomicroscope w/ calibration |
| Pericardial Edema | Pericardial area (µm²) | 15,000 ± 2,000 µm² (2 dpf) | ImageJ analysis | |
| Physiological | Heart Rate | Beats per minute (bpm) | 150 ± 20 bpm (2 dpf, 28°C) | High-speed video microscopy |
| Touch Response | Latency to movement (ms) | < 100 ms | High-speed camera (>500 fps) | |
| Behavioral | Locomotor Activity | Distance moved (mm/10 min) | 500 ± 150 mm (6 dpf, light) | Automated tracking system (DanioVision) |
| Visual Motor Response | Activity burst post-light off (% increase) | 200-300% increase | EthoVision XT software |
Table 2: Common Behavioral Assays in Adult Zebrafish
| Assay Name | Stimulus/Paradigm | Primary Readouts | Implicated Neural Circuits |
|---|---|---|---|
| Novel Tank Dive Test | Novel environment | Bottom dwelling time, freezing bouts, vertical exploration | Anxiety, stress response |
| Social Interaction | Mirror or conspecific | Interaction time, distance to stimulus | Social reward, aggression |
| Startle Response | Acoustic/vibrational tap | C-bend latency, max velocity, habituation | Sensory-motor integration |
Experimental Protocols
Protocol 1: High-Throughput Larval Morphology & Viability Screen
- Materials: CRISPR F0 mosaic mutants or F1/F2 stable mutants, wild-type siblings, egg water, 96-well plates, stereomicroscope.
- Procedure: At 24 hpf, array single embryos per well. Anesthetize with 0.02% tricaine at 48, 72, 96, and 120 hpf.
- Image Capture: Capture standardized dorsal and lateral view images using an automated microscope.
- Analysis: Use automated image analysis (e.g., CellProfiler) to measure body length, eye area, pericardial area, and yolk sac extension. Normalize all values to the plate's wild-type controls.
- Scoring: Assign phenotypic severity scores (0=wild-type, 1=mild, 2=severe). Perform statistical analysis (e.g., one-way ANOVA) comparing mutant to control groups (N≥30 per genotype).
Protocol 2: Larval Heart Rate and Rhythm Analysis
- Materials: 48-72 hpf larvae, 3% methylcellulose, tricaine (0.02%), compound microscope with high-speed camera.
- Procedure: Embed larvae laterally in methylcellulose on a glass slide. Acclimate for 5 min.
- Recording: Record a 30-second video of the heart at >100 frames per second under transmitted light.
- Analysis: Import video into software (e.g, Fiji/ImageJ with ROI manager). Count atrial and ventricular contractions over three 10-second intervals. Calculate mean heart rate (bpm) and atrioventricular interval.
- Statistical Comparison: Compare mutant heart rate and rhythm variability to sibling controls using a Student's t-test.
Protocol 3: Visual-Motor Response (VMR) Behavioral Assay
- Materials: 5-7 dpf larvae, 48-well plate, infrared-backlit behavioral rig, infrared camera, EthoVision XT.
- Acclimation: Transfer one larva per well in 500 µL egg water. Place plate in rig and acclimatize in dark for 30 min.
- Stimulus Paradigm: Program light cycle: 50 min dark, 10 min light, 10 min dark, 10 min light, 10 min dark.
- Data Acquisition: Track locomotor activity (distance moved, velocity) at 25-30 Hz throughout.
- Analysis: Calculate the average distance moved per minute. The key metric is the hyperactivity burst in the 1-2 seconds immediately following light-to-dark transitions. Compare the integrated activity of mutants versus controls during this window.
Diagrams
Title: Tiered Phenotypic Screening Workflow
Title: Neural Pathway for Visual Motor Response
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Zebrafish Phenotypic Characterization
| Item Name | Supplier Examples | Function in Phenotyping |
|---|---|---|
| Tricaine Methanesulfonate (MS-222) | Sigma-Aldrich, Pentair | Reversible anesthetic for immobilizing larvae/adults during imaging and physiological measurements. |
| Phenylthiourea (PTU) | Sigma-Aldrich | Tyrosinase inhibitor used to prevent pigment formation (0.003%) for enhanced optical clarity in imaging. |
| Methylcellulose (3%) | Sigma-Aldrich | Viscous solution for embedding and positioning larvae for high-resolution microscopy (e.g., heart imaging). |
| DanioVision Observation Chamber | Noldus Information Technology | Integrated system for controlled behavioral experiments with precise stimulus delivery and tracking. |
| EthoVision XT Software | Noldus Information Technology | Video tracking software for automated, high-throughput quantification of locomotion and behavior. |
| Tg(fli1a:EGFP) Transgenic Line | ZFIN Repository | Labels vascular endothelial cells, enabling in vivo visualization of vasculature development and integrity. |
| CellProfiler Image Analysis Software | Broad Institute | Open-source software for automated, quantitative analysis of morphological features from image datasets. |
| Fluorescent Microspheres | Thermo Fisher Scientific | Used for injection to assess cardiac output, vascular flow, or phagocytic cell function. |
This application note details the experimental and analytical workflows for leveraging CRISPR-Cas9 gene knockout in zebrafish to generate translational insights for mammalian biology and human disease. Positioned within a broader thesis on zebrafish CRISPR protocols, it emphasizes robust phenotyping and cross-species data integration to validate drug targets and dissect conserved genetic pathways.
Key Quantitative Comparisons: Zebrafish vs. Mammalian Systems
Table 1: Comparative Metrics for Gene Knockout Studies
| Metric | Zebrafish Model | Mouse Model | Human Cell Line/Organoid | Informing Insight |
|---|---|---|---|---|
| Generation Time | 3-4 months (F2 homozygous) | 9-12 months | N/A (clonal selection) | Accelerates initial in vivo target validation. |
| Embryonic Development | Externally visible, 24-120 hpf | In utero, 19-21 days | N/A | Enables real-time, high-resolution imaging of developmental phenotypes. |
| Conservation with Human Genes | ~70% (82% for disease-associated) | ~85% | 100% | High predictive value for essential biological pathways. |
| Typical N for Phenotyping | 50-200 embryos/group | 10-20 animals/group | 3-12 replicates | Enables high-powered statistical analysis of complex phenotypes. |
| Cardiovascular Function Readout | Heart rate, ejection fraction (via digital microscopy) | Echocardiography, MRI | iPSC-derived cardiomyocyte assays | Conserved physiology allows direct functional translation. |
| Neurobehavioral Assays | Touch response, locomotor activity, light/dark preference | Open field, rotarod, Morris water maze | Neuronal activity mapping (MEA) | Conserved neural circuits inform behavioral and neuropsychiatric disorders. |
Core Protocol: From Zebrafish Knockout to Mammalian Validation
Protocol 1: CRISPR-Cas9 Knockout and Primary Phenotypic Screening in Zebrafish
Objective: Generate and characterize a germline knockout of a target gene to assess its essential function.
Materials & Reagents (Research Reagent Solutions):
- CRISPR-Cas9 Components: Alt-R S.p. Cas9 Nuclease V3 (IDT) – High-specificity nuclease; Alt-R CRISPR-Cas9 tracrRNA & target-specific crRNA (IDT) – For ribonucleoprotein (RNP) complex formation.
- Microinjection Setup: Pneumatic Picopump (World Precision Instruments); borosilicate glass needles; 1x Danieau buffer.
- Genotyping: HotStarTaq Plus DNA Polymerase (Qiagen) – Robust PCR for genotyping; T7 Endonuclease I or Surveyor Mutation Detection Kit (IDT) – For indel detection; Sanger Sequencing.
- Phenotyping: Tricaine methanesulfonate (MS-222) – Anesthetic; Methyl cellulose – For embryo immobilization; In situ hybridization kit (Roche) – For gene expression analysis.
Methodology:
- Target Design: Design crRNAs against the first coding exon(s) of the zebrafish ortholog using established tools (e.g., CHOPCHOP). Select two targets to maximize knockout efficiency.
- RNP Complex Preparation: Reconstitute crRNA and tracrRNA to 100 µM. Mix 1 µL of each, heat to 95°C for 5 min, and cool. Combine 2 µL of RNA duplex, 1 µL of Alt-R Cas9 (61 µM), and 7 µL of nuclease-free water. Incubate 20 min at RT.
- Microinjection: Load RNP complex (~1 nL) into the yolk or cell of 1-cell stage embryos. Raise injected (F0) founders to adulthood.
- Founder Screening: Outcross F0 adults to wild-types. Genotype 20+ F1 progeny per clutch via PCR/HRMA or T7E1 assay on pooled embryo DNA to identify germline-transmitting founders.
- Establishing Stable Line: Raise genotyped positive F1 fish. Intercross heterozygous (F1) carriers to generate homozygous F2 mutants.
- Primary Phenotyping: Conduct blinded assessment of F2 mutants vs. siblings at 24, 48, 72, and 120 hours post-fertilization (hpf). Record viability, morphology, heart rate, circulation, and touch-evoked escape response.
Protocol 2: Cross-Species Phenotypic Alignment & Pathway Analysis
Objective: Systematically compare knockout phenotypes in zebrafish with data from murine models or human cell assays to identify conserved pathways.
Materials & Reagents (Research Reagent Solutions):
- Pathway Analysis Software: Ingenuity Pathway Analysis (IPA, Qiagen) – For cross-species pathway mapping; Zebrafish Information Network (ZFIN) – For ontology-based phenotype queries.
- Immunohistochemistry: Target-specific cross-reactive antibodies (e.g., from Abcam); Fluorescent-conjugated secondary antibodies.
- qPCR Analysis: TRIzol Reagent (Thermo Fisher) – RNA isolation; iScript cDNA Synthesis Kit (Bio-Rad); SsoAdvanced Universal SYBR Green Supermix (Bio-Rad).
Methodology:
- Phenotype Ontology Mapping: Annotate observed zebrafish phenotypes using Zebrafish Phenotype Ontology (ZPO) terms.
- Orthology Check: Verify human and mouse orthologs using Ensembl Compara. Perform reciprocal BLAST and check synteny.
- Literature Mining & Data Integration: Query databases (OMIM, MGI) for known phenotypes in mammalian models with disruptions in the orthologous gene.
- Conserved Pathway Interrogation:
- Isolate RNA from zebrafish mutant and control tissues (e.g., whole embryo or dissected organ).
- Perform qPCR for downstream genes in the hypothesized conserved pathway.
- Compare expression changes with RNA-seq or qPCR data from corresponding mouse KO models or human KO cell lines (from sources like DepMap).
- Functional Rescue with Human Gene: Clone the human wild-type cDNA into a zebrafish expression vector (e.g., pCS2+). Co-inject this mRNA with the Cas9 RNP at the 1-cell stage. Assess if the human gene rescues the mutant phenotype, confirming functional conservation.
Visualizations
Zebrafish KO to Human Biology Workflow
Conserved Growth Signaling Pathway
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for Cross-Species Knockout Studies
| Reagent / Solution | Supplier Example | Primary Function in Workflow |
|---|---|---|
| Alt-R S.p. Cas9 Nuclease V3 | Integrated DNA Technologies (IDT) | High-fidelity Cas9 enzyme for specific zebrafish knockout generation. |
| Alt-R CRISPR-Cas9 tracrRNA & crRNA | Integrated DNA Technologies (IDT) | Synthetic RNA components for forming specific RNP complexes, reducing off-target effects. |
| T7 Endonuclease I / Surveyor Kit | Integrated DNA Technologies (IDT) | Rapid detection of CRISPR-induced indel mutations in genotyping steps. |
| TRIzol Reagent | Thermo Fisher Scientific | Reliable RNA isolation from zebrafish embryos/tissues for downstream transcriptomic analysis. |
| iScript cDNA Synthesis Kit | Bio-Rad Laboratories | Efficient cDNA synthesis from low-quantity zebrafish RNA samples. |
| Cross-Reactive Antibody (e.g., anti-pAkt) | Cell Signaling Technology | Validates conserved pathway activation/inhibition across zebrafish and mammalian tissue samples. |
| Ingenuity Pathway Analysis (IPA) | Qiagen Bioinformatics | Software for cross-species molecular pathway mapping and data integration. |
| pCS2+ Expression Vector | Addgene | Standard plasmid for synthesizing mRNA for rescue experiments with human genes. |
Within the broader thesis on CRISPR-Cas9 gene knockout protocols in zebrafish, this application note details the translational pipeline from genetic manipulation to preclinical drug discovery. Zebrafish knockout (KO) lines, generated via standardized CRISPR-Cas9 injection and screening protocols, serve as powerful in vivo platforms for validating novel therapeutic targets and screening for compound-induced toxicities. Their high genetic and physiological conservation with humans, coupled with optical transparency and rapid development, enables medium-throughput, quantitative phenotypic analysis.
Application Note 1:In VivoTarget Validation Using Genetic Knockouts
Objective: To confirm that modulation of a hypothesized target gene produces a therapeutic-relevant phenotype, thereby de-risking it for further drug development.
Protocol: Phenotypic Rescue for Target Validation
- KO Line Generation: Generate a stable homozygous knockout line for the gene of interest (Target X) using established CRISPR-Cas9 protocols (gRNA design, microinjection, F0 screening, and incrossing to establish F2 homozygotes).
- Phenotypic Characterization: Quantitatively assess the KO phenotype using high-content imaging and behavioral assays (e.g., cardiac function, angiogenesis, neuronal development, or disease-model specific endpoints).
- Pharmacological/Genetic Rescue:
- Small Molecule Rescue: Expose Target X KO embryos from 0-3 days post-fertilization (dpf) to a known agonist or activator of the pathway downstream of Target X. A positive rescue towards wild-type (WT) phenotype validates Target X as a key nodal point.
- mRNA Rescue: Co-inject in vitro-transcribed human Target X mRNA with Cas9/gRNA at the 1-cell stage. Assessment of phenotype mitigation confirms target specificity and demonstrates functional conservation.
- Data Analysis: Compare endpoint measurements between WT, KO, and rescue groups using ANOVA with post-hoc tests.
Key Data Output (Example: Angiogenesis Target): Table 1: Quantitative Phenotypic Analysis of *vegfr2 Knockout and Rescue*
| Genotype/Treatment | Intersegmental Vessel Count (Mean ± SD) | Vessel Length (µm, Mean ± SD) | Rescue Significance (p-value vs. KO) |
|---|---|---|---|
| WT | 52.3 ± 3.1 | 298.5 ± 22.4 | - |
| vegfr2 KO | 12.7 ± 5.8 | 85.3 ± 31.6 | - |
| KO + VEGFA (50 ng/mL) | 45.6 ± 6.2 | 265.8 ± 28.9 | <0.001 |
Application Note 2: Systematic Toxicity and Off-Target Screening
Objective: To utilize specific organelle- or pathway-targeted KO lines as sensitive biosensors for predicting compound toxicity mechanisms.
Protocol: Liver Toxicity (Steatosis) Screening Workflow
- Biosensor Lines: Utilize a transgenic liver fluorescent reporter line (e.g., Tg(fabp10a:DsRed)) crossed into a knockout background for a key metabolic gene (e.g., mtp, microsomal triglyceride transfer protein).
- Compound Exposure:
- Array 5 dpf larvae in 96-well plates (n=10 larvae/well).
- Treat with a dilution series of the candidate drug (1 µM - 100 µM) or DMSO control. Include a positive control (e.g., 250 µM acetaminophen).
- Expose for 24-48 hours.
- Endpoint Analysis:
- Viability & Malformations: Score mortality and gross morphology.
- Liver Size & Function: Image fluorescence to quantify liver size. Perform a fluorescent triglyceride dye (e.g., Nile Red) uptake assay.
- Biochemical Assay: Homogenize larvae in pools and measure ALT/AST levels via colorimetric assay kits.
- Hit Definition: A compound is flagged for hepatotoxicity if it causes significant (p<0.05) enhancement of steatosis in the mtp KO background compared to WT at non-teratogenic concentrations.
Key Data Output (Example): Table 2: Hepatotoxicity Profiling of Candidate Drug Y in Sensitized *mtp KO Line*
| Genotype | [Drug Y] (µM) | Viability (%) | Liver Area (µm², Mean ± SD) | Nile Red Intensity (Mean ± SD) | ALT (U/L) |
|---|---|---|---|---|---|
| WT | 0 (DMSO) | 100 | 12500 ± 1500 | 1050 ± 230 | 22 ± 4 |
| WT | 10 | 98 | 11800 ± 2100 | 1200 ± 310 | 28 ± 7 |
| mtp KO | 0 (DMSO) | 95 | 14500 ± 1800 | 4500 ± 850 | 55 ± 12 |
| mtp KO | 10 | 65 | 21000 ± 3200 | 12500 ± 2200 | 180 ± 45 |
Visualizations
Diagram 1: Target validation and screening workflow.
Diagram 2: Toxicity screening logic with sensitized KOs.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Zebrafish KO-based Drug Discovery Applications
| Reagent / Solution | Function & Application in Protocol | Example Product / Note |
|---|---|---|
| CRISPR-Cas9 Components | Generation of knockout lines. | High-fidelity Cas9 protein, gene-specific gRNA (synthesized or in vitro transcribed), PCR-based genotyping kits. |
| Embryo Medium & Micronjection Supplies | Maintenance and manipulation of embryos. | E3 medium, glass capillary needles, micromanipulator, phenol red tracer. |
| Automated Imaging System | High-content phenotypic screening. | Systems with fluidics for 96-well plates (e.g., ImageXpress, VAST Bioimager) coupled with analysis software (e.g., ImageJ, Fiji). |
| Fluorescent Reporter Lines | Visualizing organ-specific effects. | Tg(fabp10a:DsRed) (liver), Tg(myl7:EGFP) (heart), Tg(fli1a:EGFP) (vasculature). |
| Pathway-Specific Biosensor Lines | Reporting on pathway activity. | TGF-β, Wnt, or Apoptosis reporter transgenic lines in WT/KO backgrounds. |
| Compound Libraries | Pharmacological screening. | FDA-approved drug libraries, targeted small-molecule collections. Dissolved in DMSO for aqueous dilution. |
| Specialized Stains & Dyes | Labeling cellular structures/processes. | Nile Red (lipid droplets), Acridine Orange (apoptosis), BODIPY (oxidative stress), Alcian Blue (cartilage). |
| Microplate Reader-Compatible Assay Kits | Quantitative biochemical readouts from larvae homogenates. | ALT/AST activity, ATP content, caspase-3/7 activity, total glutathione. |
| Statistical Analysis Software | Data analysis and visualization. | GraphPad Prism, R, Python (Pandas, SciPy). |
Conclusion
CRISPR-Cas9 has revolutionized functional genomics in zebrafish, offering a rapid, cost-effective, and highly tractable system for generating precise gene knockouts. Success hinges on meticulous sgRNA design, optimized microinjection techniques, systematic troubleshooting, and rigorous multi-layered validation. As protocols continue to be refined with improved Cas9 variants and delivery methods, zebrafish knockouts will play an increasingly pivotal role in bridging the gap between genetic discovery and clinical application. Future directions include large-scale multiplexed knockouts, tissue-specific editing, and the integration of knockout models with high-throughput drug screening platforms, solidifying the zebrafish's position as an indispensable model for biomedical research and therapeutic development.