CRISPR Screen Data Normalization: Essential Methods for Robust Analysis in 2024
This comprehensive guide explores the critical role of data normalization in CRISPR screen analysis.
CRISPR Screen Data Normalization: Essential Methods for Robust Analysis in 2024
Abstract
This comprehensive guide explores the critical role of data normalization in CRISPR screen analysis. Tailored for researchers, scientists, and drug development professionals, it provides a foundational understanding of normalization concepts, details step-by-step methodologies for different screen types (e.g., dropout, enrichment), addresses common pitfalls and troubleshooting strategies, and offers a comparative analysis of validation techniques. The article empowers users to select and implement optimal normalization strategies to extract reliable biological insights from CRISPR screening experiments, enhancing the reproducibility and impact of their functional genomics research.
Understanding CRISPR Screen Data Normalization: Why It's the Cornerstone of Reliable Analysis
Within the broader research on CRISPR screen data normalization methods, effective normalization is not a mere preprocessing step but the foundational process for distinguishing true biological signal from technical and biological noise. Functional genomics, particularly genome-wide CRISPR knockout or perturbation screens, generates complex datasets where observed read counts are confounded by factors like sequencing depth, gRNA library composition, and cell-specific fitness effects. This document details application notes and protocols for implementing and validating normalization methods critical for robust hit identification in therapeutic target discovery.
The primary objective is to adjust raw gRNA or gene-level counts to enable fair comparison across samples and conditions. Key noise sources include:
- Library Size: Differences in total sequencing reads between samples.
- gRNA Efficiency: Variable cutting efficiency among gRNAs targeting the same gene.
- Batch Effects: Technical variations from different experimental runs.
- Cell Growth Effects: Non-uniform proliferation rates influenced by essential gene knockout.
- PCR Amplification Bias: Uneven amplification during library preparation.
Quantitative Comparison of Normalization Methods
The performance of normalization methods is typically evaluated using benchmark datasets with known essential and non-essential genes (e.g., DepMap core fitness genes). Key metrics include precision-recall AUC, false discovery rate (FDR) control, and robustness across cell lines.
Table 1: Comparison of Common CRISPR Screen Normalization Methods
| Method | Core Principle | Best For | Key Assumption | Software/Tool |
|---|---|---|---|---|
| Median-of-Ratios | Scales counts based on the median of gene-wise ratios to a reference sample. | Basic correction for sequencing depth. | Most genes are not differentially enriched/depleted. | DESeq2, MAGeCK |
| Total Count (CPM) | Normalizes to counts per million mapped reads. | Simple, quick assessment. | Total library size is the main bias. | Basic R/Python |
| RRA (Robust Rank Aggregation) | Ranks gRNAs within a sample to aggregate gene-level scores; reduces outlier impact. | Screens with strong positive/negative selection. | The rank of gRNAs is more reliable than raw counts. | MAGeCK, MAGeCK-VISPR |
| Control Gene (e.g., Non-Targeting) | Uses a set of non-targeting or safe-harbor targeting gRNAs as a neutral reference distribution. | Accounting for sequence-specific & cell-type specific noise. | Control gRNAs capture the null distribution of fitness effects. | BAGEL2, CERES |
| CERES | Jointly estimates gene knockout effect and a cell line-specific nuisance factor. | Pooled screens across many cell lines (pan-cancer). | Confounding factors are shared across genes in a cell line. | DepMap (Avana libraries) |
Table 2: Performance Metrics on DepMap Benchmark (Hypothetical Data) Performance evaluated using Precision-Recall AUC for recovering known essential genes.
| Method | HAP1 Cell Line (AUC) | A375 Cell Line (AUC) | HeLa Cell Line (AUC) | Median FDR (%) |
|---|---|---|---|---|
| Total Count | 0.72 | 0.65 | 0.68 | 12.5 |
| Median-of-Ratios | 0.81 | 0.78 | 0.79 | 8.2 |
| RRA | 0.88 | 0.82 | 0.85 | 5.5 |
| Control Gene (BAGEL2) | 0.92 | 0.90 | 0.91 | 3.8 |
| CERES | 0.94 | 0.93 | 0.92 | 2.9 |
Experimental Protocols
Protocol 4.1: Standard Workflow for CRISPR Screen Data Normalization & Analysis
Objective: Process raw FASTQ files from a viability screen to a list of significant hit genes. Duration: 2-3 days (computational). Reagents/Software: High-performance computing environment, MAGeCK (version 0.5.9+), R/Bioconductor.
Procedure:
- gRNA Quantification:
- Align sequencing reads (
sample.fastq) to the reference gRNA library (library.txt) usingmageck count. - Command:
mageck count -l library.txt -n sample_output --sample-label Sample1 --fastq sample.fastq.gz - Output: A raw count table (
sample_output.count.txt).
- Align sequencing reads (
Normalization & Test:
- Perform median normalization and calculate gene-level beta scores using the robust rank aggregation (RRA) algorithm via
mageck test. - Command:
mageck test -k sample_output.count.txt -t Treatment_Sample -c Control_Sample -n test_output --norm-method total - Specify
--control-geneif a list of non-essential genes is provided for alternative normalization.
- Perform median normalization and calculate gene-level beta scores using the robust rank aggregation (RRA) algorithm via
Hit Calling & FDR Control:
- Analyze output file
test_output.gene_summary.txt. Genes with a positive selection p-value < 0.05 and FDR < 0.1 are candidate essential hits (depleted in treatment). Genes with a negative selection p-value < 0.05 and FDR < 0.1 are candidate resistance hits (enriched).
- Analyze output file
Visualization:
- Generate rank plots and gRNA read count distributions using
mageck visualor custom R scripts (ggplot2).
- Generate rank plots and gRNA read count distributions using
Protocol 4.2: Validation of Normalization Using Positive Control Genes
Objective: Empirically assess normalization quality by measuring the separation between known essential and non-essential gene distributions. Duration: 1 day (computational).
Procedure:
- Obtain a curated list of core essential genes (e.g., from DepMap) and a list of non-essential genes (e.g., from Gene Ontology terms for extracellular processes).
- Run your CRISPR screen analysis pipeline (as in Protocol 4.1) to generate normalized gene scores (e.g., log2 fold-change or beta score).
- Extract the normalized scores for the essential and non-essential gene sets.
- Perform a Wilcoxon rank-sum test to confirm the scores for essential genes are significantly lower (depleted) than non-essential genes.
- Calculate the effect size (e.g., Cohen's d). A larger absolute effect size indicates better normalization and signal separation.
- Plot the distributions as violin or box plots for qualitative assessment.
Visualization Diagrams
Normalization Pipeline to Remove Sequential Noise
CRISPR Screen Workflow from Lab to Analysis
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for CRISPR Screen Normalization & Validation
| Item | Function in Normalization Context | Example/Provider |
|---|---|---|
| Non-Targeting gRNA Library | Provides a set of control guides that define the null distribution of fitness effects. Critical for control-based normalization methods. | Synthego, Horizon Discovery, Addgene (e.g., pLCKO non-targeting library) |
| Benchmark Essential Gene Set | Gold-standard list of pan-essential genes used to validate normalization method performance and calculate AUC metrics. | DepMap Core Fitness Genes (CEGv2), Hart et al. (2015) gene list. |
| CRISPR Analysis Software Suite | Tools that implement various normalization algorithms (total count, median, RRA, control-based). | MAGeCK, BAGEL2, PinAPL-Py, CRISPRcleanR. |
| Cell Lines with Defined Fitness | Cell lines with well-characterized essential/non-essential genes for method benchmarking. | HAP1 (near-haploid), K562, A375. |
| Synthetic Lethal/Positive Control gRNAs | gRNAs targeting known essential genes (e.g., RPA3) used as internal controls to monitor screen dynamic range and normalization efficacy. | Custom synthesis from IDT or Twist Bioscience. |
| Spike-in DNA/RNA Controls | External controls added during library prep to potentially correct for amplification and sequencing batch effects. | ERCC RNA Spike-In Mix (Thermo Fisher). |
Within the broader thesis on CRISPR screen data normalization methods, this document details the core technical challenges that necessitate robust normalization. CRISPR screening generates high-dimensional functional genomics data, where raw sequencing counts are confounded by non-biological noise. Effective normalization is not merely a preprocessing step but a foundational correction that isolates true gene essentiality signals from artifacts. The following Application Notes and Protocols focus on three pervasive challenges: batch effects, library-specific biases, and disparities in sequencing depth.
Batch Effects
Batch effects are systematic technical variations introduced when samples are processed in different experimental batches (e.g., different days, sequencing lanes, or reagent lots). They can confound biological signals and lead to false conclusions.
Protocol: Identifying and Correcting for Batch Effects via Negative Controls
Objective: To diagnose and mitigate batch effects using non-targeting sgRNA controls.
Materials: Processed read counts from a CRISPR screen conducted across multiple batches.
Procedure:
1. Data Aggregation: Compile raw sgRNA count tables for all samples and batches.
2. Control Selection: Isolate the read counts for the set of non-targeting control sgRNAs present in the library.
3. PCA Visualization: Perform Principal Component Analysis (PCA) on the log-transformed counts of the control sgRNAs only.
4. Batch Diagnosis: Visualize the first two principal components. Clustering of samples by batch rather than biological condition indicates a strong batch effect.
5. Normalization Application: Apply a batch correction method. A common approach is using the removeBatchEffect function from the R limma package, using the control sgRNA data to estimate the batch-associated variation.
6. Validation: Re-run PCA on the normalized control sgRNA counts. Successful correction is indicated by the mixing of samples from different batches.
Table 1: Impact of Batch Correction on sgRNA Replicate Correlation
| Sample Pair (Biological Replicates) | Correlation (Raw Counts) | Correlation (Batch-Corrected) | Method Used |
|---|---|---|---|
| Rep1 (Batch A) vs. Rep2 (Batch B) | 0.72 | 0.91 | limma |
| Rep1 (Batch A) vs. Rep3 (Batch C) | 0.65 | 0.89 | limma |
| Average Improvement | +0.23 |
Title: Batch Effect Diagnosis and Correction Workflow
Library Biases
Library biases refer to systematic differences in sgRNA abundance and functionality inherent to the design of the sgRNA library itself. These include variations in DNA synthesis efficiency, genomic integration rates, and on-target cutting efficiency.
Protocol: Normalizing for Library-Specific Bias Using Total Read Scaling Objective: To adjust counts for global differences in sgRNA representation and recovery. Materials: Raw FASTQ files and the reference sgRNA library file. Procedure: 1. Read Alignment: Align sequencing reads to the reference sgRNA library using a short-read aligner (e.g., Bowtie 2, BWA). 2. Raw Count Generation: Tally the number of reads uniquely mapped to each sgRNA identifier. 3. Calculate Scaling Factors: For each sample, compute a size factor. The Median-of-Ratios method (as in DESeq2) is widely used: a. Create a pseudo-reference sample by taking the geometric mean of each sgRNA count across all samples. b. For each sample, compute the ratio of each sgRNA's count to the pseudo-reference count. c. The scaling factor for the sample is the median of these ratios (excluding sgRNAs with zero counts in either sample). 4. Apply Normalization: Divide the raw counts for each sgRNA in a sample by that sample's scaling factor. This yields normalized counts (often as "counts per million" or analogous). 5. Quality Assessment: Plot the distribution of log2-normalized counts across samples. Distributions should align centrally post-normalization.
Table 2: Example of Scaling Factors Across Samples with Varying Library Complexity
| Sample ID | Total Raw Reads (M) | Median-of-Ratios Scaling Factor | Normalized Effective Depth (M) |
|---|---|---|---|
| S1 | 45.2 | 1.05 | 43.0 |
| S2 | 68.7 | 0.78 | 88.1 |
| S3 | 32.5 | 1.45 | 22.4 |
Sequencing Depth
Differences in total sequencing depth between samples create technical variation in sgRNA count magnitude, which can obscure biological differences in dropout.
Protocol: Depth Normalization and Essential Gene Calling with MAGeCK
Objective: To compare gene essentiality scores across screens of differing sequencing depths.
Materials: Normalized sgRNA count tables from multiple screens/conditions.
Procedure:
1. Input Preparation: Prepare a count matrix of normalized sgRNA counts (from Protocol 2) for all samples.
2. Run MAGeCK MLE: Use the Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) MLE algorithm to account for sequencing depth and sgRNA variance.
Command: mageck mle -k sample_count_table.txt -d design_matrix.txt -n output_prefix
3. Parameterization: The design matrix encodes sample relationships. The algorithm internally models the mean-variance relationship of sgRNAs, down-weighting noisy sgRNAs and implicitly normalizing for depth via its negative binomial model.
4. Output Interpretation: Key outputs include gene beta scores (log-fold-change) and p-values. A positive beta score indicates gene enrichment in a condition; a negative score indicates essentiality (depletion).
5. Benchmarking: Compare the ranked list of essential genes from a deep-sequenced sample versus a shallow one before and after MAGeCK normalization. The rank order should stabilize post-normalization.
Table 3: Gene Ranking Stability Before/After Depth Normalization
| Gene | Rank in Deep Sample (Raw) | Rank in Shallow Sample (Raw) | Rank in Deep Sample (MAGeCK) | Rank in Shallow Sample (MAGeCK) |
|---|---|---|---|---|
| Gene A | 1 | 15 | 1 | 2 |
| Gene B | 2 | 45 | 3 | 5 |
| Gene C | 3 | 8 | 4 | 3 |
| Spearman Correlation vs. Deep Sample | - | 0.71 (Raw) | - | 0.97 (MAGeCK) |
Title: Sequencing Depth Normalization via MAGeCK MLE
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for CRISPR Screen Normalization & Analysis
| Item | Function in Context | Example/Supplier |
|---|---|---|
| Validated Non-targeting Control sgRNA Library | Serves as a null baseline for identifying batch effects and technical noise. | Horizon Discovery, Sigma-Aldrich |
| Bowtie 2 Aligner | Aligns sequencing reads to the sgRNA reference library with high speed and accuracy for raw count generation. | Open Source (http://bowtie-bio.sourceforge.net/bowtie2) |
| DESeq2 R/Bioconductor Package | Provides the Median-of-Ratios method for library size normalization and differential analysis. | Bioconductor |
| MAGeCK Software Suite | Comprehensive toolkit for normalizing count data, calling essential genes, and correcting for multiple confounders including depth. | Open Source (https://sourceforge.net/p/mageck) |
| limma R/Bioconductor Package | Contains robust functions for removing batch effects from high-dimensional data. | Bioconductor |
| High-Complexity sgRNA Library | A well-designed library (e.g., Brunello, Brie) with multiple sgRNAs per gene, enabling robust internal normalization. | Addgene (https://www.addgene.org) |
| Spike-in Control (e.g., ePCR) | Exogenous oligonucleotides added pre-PCR to normalize for amplification biases across samples. | Custom synthesis (IDT, etc.) |
Application Notes
Within the framework of CRISPR screen data normalization research, the precise definition and calculation of key metrics form the foundational layer for accurate biological interpretation. These metrics transform raw sequencing data into quantifiable measures of gene function and fitness.
1. Read Counts: These are the raw, integer counts of sequencing reads uniquely aligned to each sgRNA in a sample. They represent the starting point for all analyses but are subject to technical noise from variations in sequencing depth and PCR amplification.
2. sgRNA Abundance: This is a normalized measure of sgRNA representation within a library, typically derived from read counts. It corrects for differences in total library size between samples, enabling direct comparison. Common normalization methods include:
- Counts Per Million (CPM): Scales reads by the total library size.
- Median-of-Ratios (e.g., DESeq2): Estimates size factors based on the assumption that most sgRNAs are not differentially abundant.
- Trimmed Mean of M-values (TMM): Removes extreme values to calculate scaling factors.
3. Gene-Level Scores: These scores aggregate data from multiple sgRNAs targeting the same gene to infer a gene's effect on the phenotype. This step increases statistical power and mitigates sgRNA-level noise and off-target effects. Common aggregation methods include:
- Robust Ranking Aggregation (RRA): Ranks sgRNAs by their enrichment/depletion significance across replicates and aggregates ranks.
- STARS: Uses a permutation-based method to assess the reproducibility of high-ranking sgRNAs per gene.
- MAGeCK: Employs a negative binomial model or a robust rank aggregation algorithm (MAGeCK-RRA) to test for significant gene-level selection.
Quantitative Comparison of Common Normalization & Scoring Methods
| Metric/Method | Primary Function | Key Input | Key Output | Advantages | Limitations |
|---|---|---|---|---|---|
| Total Read Count | Raw data quantification | FASTQ files | Integer counts per sgRNA | Simple, unbiased starting point | Highly dependent on sequencing depth |
| CPM | Library size normalization | Raw read counts | Normalized abundance per 1M reads | Intuitive, computationally simple | Sensitive to highly abundant sgRNAs skewing totals |
| DESeq2 Median-of-Ratios | Library size & composition normalization | Raw read counts | Normalized abundance (continuous) | Robust to composition bias, handles replicates well | Assumes most sgRNAs are non-DE; can be conservative |
| MAGeCK (beta score) | Gene-level essentiality scoring | Normalized counts (T0, Tx) | β score (log2 fold change) & p-value | Integrates variance modeling, handles multiple timepoints | Complex model, requires understanding of parameters |
| RRA (from MAGeCK or BAGEL) | Gene-ranking & significance | sgRNA fold changes/ranks | Rank & FDR per gene | Non-parametric, robust to outliers | May lose information about effect size magnitude |
Experimental Protocols
Protocol 1: From FASTQ to Normalized sgRNA Abundance Matrix
Objective: Process raw sequencing files to generate a normalized count matrix for downstream analysis.
Materials: (See The Scientist's Toolkit) Software: cutadapt, Bowtie2, MAGeCK-count, R with DESeq2.
Procedure:
- Demultiplex and Trim Adapters: Use
cutadaptto remove constant adapter sequences and sample barcodes.cutadapt -a [ADAPTER_SEQ] -o output.fastq input.fastq
- Align Reads to sgRNA Library Reference: Map trimmed reads to a FASTA file of all expected sgRNA sequences using
Bowtie2in end-to-end, sensitive mode.bowtie2 -x sgRNA_lib_index -U input_trimmed.fastq -S output.sam
- Generate Raw Count Table: Use
MAGeCK countor a custom script to count alignments per sgRNA per sample from the SAM/BAM file.mageck count -l library.csv -n output_count --sample-label Sample1 [--fastq sample1.fastq]
- Normalize for Library Size: In R, using the raw count matrix (
counts), apply the DESeq2 median-of-ratios method.
Protocol 2: Calculating Gene-Level Scores Using MAGeCK-RRA
Objective: Aggregate sgRNA-level fold changes to identify significantly enriched or depleted genes.
Materials: Normalized count matrix, sgRNA-to-gene annotation file. Software: MAGeCK (version 0.5.9+).
Procedure:
- Prepare Input Files: Ensure you have:
- A count table file (
counts.txt) with rows as sgRNAs and columns as samples. - A sample annotation file specifying which columns are treatment (Tx) and control (T0).
- An sgRNA library file linking each sgRNA to its target gene.
- A count table file (
- Run MAGeCK RRA Test: Execute the
mageck testcommand with the RRA algorithm. - Interpret Output: Key files include:
output_results.gene_summary.txt: Contains gene-level β scores (log2 fold change), p-values, and FDR. Genes with positive β are depleted in the treatment; negative β indicates enrichment.
Visualizations
CRISPR Screen Data Analysis Workflow
Factors Influencing Key CRISPR Screen Metrics
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in CRISPR Screen Metrics Analysis |
|---|---|
| Validated sgRNA Library Plasmid Pool (e.g., Brunello, GeCKO) | Provides the starting genetic material with known sequences, essential for creating the alignment reference and annotation files. |
| Next-Generation Sequencing Kit (Illumina NovaSeq, MiSeq) | Generates the raw FASTQ data. Read depth and quality directly impact the robustness of read count data. |
| PCR Amplification Primers with Illumina Adapters | Amplifies sgRNA representation from genomic DNA for sequencing. Must be optimized to minimize amplification bias affecting count distribution. |
| sgRNA Library Reference FASTA File | Contains the DNA sequence of every sgRNA in the library. Critical for the alignment step to assign reads correctly. |
| Negative Control sgRNAs (e.g., Targeting Non-Human Genome) | Used to model the null distribution of fold changes, improving false discovery rate (FDR) estimation in gene-level scoring. |
| Positive Control sgRNAs (e.g., Targeting Essential Genes) | Provide a benchmark for screen performance and normalization efficacy, confirming expected depletion in abundance metrics. |
| MAGeCK Software Suite | Comprehensive, open-source toolkit that standardizes the pipeline from count processing to gene-level scoring, ensuring reproducibility. |
| R/Bioconductor with DESeq2 & edgeR Packages | Provides industry-standard statistical frameworks for robust normalization of count data between samples. |
| BAGEL (Bayesian Analysis of Gene Essentiality) | Alternative, complementary tool for gene-level scoring that uses a gold-standard reference set of essential/non-essential genes for Bayesian classification. |
1. Introduction within CRISPR Screen Data Normalization Research This document provides application notes and experimental protocols for the normalization of data from two primary CRISPR-Cas9 screening paradigms: dropout (negative selection) and enrichment (positive selection) screens. Effective normalization is a critical component of a robust data analysis pipeline, directly impacting the accuracy of hit identification. The broader thesis posits that the distinct biological and statistical characteristics of these screen types necessitate tailored, non-interchangeable normalization strategies to control for differing sources of technical and biological variance.
2. Core Concepts and Normalization Imperatives
Dropout (Negative Selection) Screens: Aim to identify genes essential for cell fitness or survival under a given condition (e.g., standard culture, treatment with a toxin). Cells carrying sgRNAs targeting these genes are depleted from the population over time.
- Primary Need: Control for variance in initial sgRNA representation and sequencing depth. Normalization must accurately quantify depletion.
- Key Challenge: Distinguishing true lethal phenotypes from background stochastic dropout, especially at early time points or in screens with moderate effect sizes.
Enrichment (Positive Selection) Screens: Aim to identify genes whose loss confers a growth advantage or resistance to a selective pressure (e.g., drug treatment, pathogen infection). Cells with sgRNAs targeting these genes become enriched.
- Primary Need: Control for variance in screening potency and dynamic range. Normalization must accurately quantify enrichment.
- Key Challenge: Distinguishing true positive hits from passenger effects and accounting for potential bottlenecks during selection.
3. Comparative Analysis: Quantitative Data Summary
Table 1: Characteristics and Normalization Requirements of Primary CRISPR Screen Types
| Feature | Dropout / Negative Selection Screen | Enrichment / Positive Selection Screen |
|---|---|---|
| Biological Goal | Identify essential genes (e.g., viability, fitness) | Identify genes conferring resistance or advantage |
| Phenotype | Depletion of sgRNA guides over time | Enrichment of sgRNA guides over time |
| Typical Duration | Longer (e.g., 14-21 cell doublings) | Shorter, defined by selective agent |
| Key Statistical Distribution | Negative binomial (count data, overdispersion) | Often more skewed; can approach zero-inflated |
| Primary Normalization Focus | Read count scaling, control sgRNA-based correction (e.g., non-targeting, core essentials) | Fold-change calculation, variance stabilization for low-count starts |
| Major Noise Sources | Variable initial transduction, growth rate differences | Selection bottleneck strength, pre-selection library complexity |
| Common Hit Threshold | Significant negative log2 fold-change (e.g., <-2) & p-value | Significant positive log2 fold-change (e.g., >2) & p-value |
| Example Analysis Tools | MAGeCK, BAGEL, CERES | MAGeCK, edgeR, DESeq2 |
4. Experimental Protocols
Protocol 1: Standard Workflow for a Dropout Screen with Median Ratio Normalization
A. Library Transduction and Passaging
- Seed HeLa cells in a 6-well plate at 30% confluence.
- Transduce cells with the Brunello human whole-genome CRISPRko library (Addgene #73178) at an MOI of ~0.3 in the presence of 8 µg/mL polybrene.
- Select: 24 hours post-transduction, begin selection with 2 µg/mL puromycin for 72 hours.
- Harvest T0 Sample: Collect ≥ 5e6 cells, pellet, and store at -80°C for genomic DNA (gDNA) extraction. This is the reference time point.
- Passage: Maintain the remaining population, passaging every 2-3 days to keep cells in exponential growth. Harvest an equivalent cell number (≥5e6) at passages corresponding to T14 and T21 (~14 and 21 population doublings).
B. gDNA Extraction & NGS Library Preparation
- Extract gDNA from cell pellets using the QIAamp DNA Blood Maxi Kit.
- Amplify sgRNA sequences via a two-step PCR protocol.
- PCR1 (sgRNA locus amplification): Use 10 µg gDNA per sample in 8 parallel 100 µL reactions with Herculase II polymerase. Cycle conditions: 98°C 2min; 30 cycles of (98°C 20s, 60°C 20s, 72°C 30s); 72°C 3min.
- Clean up PCR1 products with AMPure XP beads.
- PCR2 (Add Illumina adaptors & indices): Use 2 µL of cleaned PCR1 product per reaction. Cycle conditions: 98°C 2min; 12 cycles of (98°C 20s, 65°C 20s, 72°C 30s); 72°C 3min.
- Clean up PCR2 products with AMPure XP beads, quantify, pool equimolar amounts, and sequence on an Illumina NextSeq 500 (75bp single-end, targeting >500 reads per sgRNA).
C. Data Normalization & Analysis (Median-of-Ratios)
- Align reads to the sgRNA library reference using
magck count. - Normalize read counts across all samples (T0, T14, T21) using a median-of-ratios method (as in DESeq2/MAGeCK) to correct for differences in total sequencing depth and gDNA amplification efficiency.
- Calculate log2 fold-changes (LFC) for each sgRNA between T0 and later time points.
- Fit a robust regression model (e.g., in MAGeCK RRA algorithm) using a set of non-targeting control sgRNAs to estimate the null distribution of dropout and identify significantly depleted genes (FDR < 0.05).
Protocol 2: Standard Workflow for an Enrichment Screen with Variance Stabilizing Transformation
A. Library Transduction and Selection
- Seed A375 cells in a 6-well plate at 30% confluence.
- Transduce with the same Brunello library as in Protocol 1, Step A2.
- Puromycin Selection: Perform selection as in Protocol 1, Step A3.
- Harvest Pre-Selection (T0) Sample: Collect cells 72 hours after puromycin selection ends (Day 0 of treatment).
- Apply Selective Pressure: Split the remaining cells and treat with 1 µM vemurafenib (PLX4032) or DMSO vehicle control. Culture for 14-21 days, replenishing drug/media every 3 days.
- Harvest Post-Selection (Tsel) Samples: Collect resistant cell populations.
B. NGS Library Preparation & Sequencing
- Perform exactly as in Protocol 1, Section B.
C. Data Normalization & Analysis (Variance Stabilization)
- Align reads using
magck count. - Normalize: For enrichment screens, variance tends to be count-dependent (higher variance at lower counts). Apply a variance-stabilizing transformation (VST) to the count data (e.g., via DESeq2's
vstfunction) before fold-change calculation. - Calculate LFCs for each sgRNA comparing Tsel (drug) to Tsel (DMSO) and to the initial T0 sample to control for baseline representation biases.
- Perform statistical testing using a model (e.g., MAGeCK MLE) that incorporates variance from both sample replicates and the T0 reference to identify significantly enriched genes (FDR < 0.05).
5. Signaling and Workflow Visualizations
Dropout Screen Workflow
Enrichment Screen Workflow
6. The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for CRISPR Screening Experiments
| Item | Function & Relevance to Normalization |
|---|---|
| Genome-Scale sgRNA Library (e.g., Brunello, GeCKO) | Defines screen's genetic space. High-quality, uniformly synthesized libraries minimize representation bias, a key pre-normalization factor. |
| Non-Targeting Control sgRNA Pool | Contains sgRNAs not targeting any genomic locus. Critical for determining the null distribution of phenotype in both dropout and enrichment screens during statistical modeling. |
| Core Essential Gene sgRNA Set | A panel of sgRNAs targeting genes universally required for cell viability. Used specifically in dropout screens as a positive control for assay performance and for normalization (e.g., in BAGEL2). |
| Puromycin (or appropriate antibiotic) | For stable selection of transduced cells, ensuring high library representation at T0, which is foundational for accurate downstream count comparison. |
| Polybrene / Hexadimethrine bromide | Enhances viral transduction efficiency, promoting uniform library representation across the cell population. |
| High-Yield gDNA Extraction Kit (e.g., QIAamp Maxi) | Consistent, high-quality gDNA recovery is vital for unbiased PCR amplification of all sgRNA templates across samples. |
| High-Fidelity PCR Polymerase (e.g., Herculase II) | Minimizes amplification bias during NGS library prep, ensuring final read counts accurately reflect initial sgRNA abundances. |
| AMPure XP Beads | For precise size selection and clean-up of PCR amplicons, removing primers and primer-dimers that skew sequencing results. |
| Illumina Sequencing Platform | Provides the quantitative count data. Sufficient sequencing depth (>500x coverage) is required to detect meaningful fold-changes, especially for depleted sgRNAs. |
Application Notes
This document provides foundational concepts and methodologies for normalizing high-throughput sequencing data from CRISPR-Cas9 knockout screens. These normalization techniques are critical for removing technical noise and systematic biases, enabling accurate identification of genes essential for cell fitness and drug-gene interactions.
Median Ratio Normalization assumes most features (sgRNAs) are non-differentially abundant. It calculates a size factor for each sample as the median of the ratios of observed counts to a pseudo-reference sample. Quantile Normalization enforces the same empirical distribution of counts across all samples, aligning quantiles. Variance Stabilizing Transformation (VST) models the mean-variance relationship in count data (where variance increases with mean) and transforms the data to stabilize variance across the dynamic range, making it more amenable to statistical testing.
These methods are essential preprocessing steps prior to downstream analysis, such as MAGeCK or DrugZ, to rank essential genes or identify sensitizing/resistance interactions.
Core Concepts & Quantitative Data
Table 1: Comparison of Normalization Methods for CRISPR Screen Data
| Method | Primary Assumption | Handles Zeros? | Preserves Magnitude? | Best For |
|---|---|---|---|---|
| Median Ratio | Majority of sgRNAs are non-hit. | Yes, uses geometric mean. | No, scales data. | Standard essential screens with moderate effect sizes. |
| Quantile | Overall sgRNA distribution is similar across samples. | Problematic; distorts zero structure. | No, forces identical distributions. | Samples with very similar phenotypes and high replicate correlation. |
| Variance Stabilizing Transform | Variance is a function of mean (Poisson/ Negative Binomial). | Yes, handled by underlying model. | No, transforms to a new scale. | Downstream linear modeling (e.g., for drug-genes screens with continuous phenotypes). |
Table 2: Impact of Normalization on Key Metrics (Simulated Data)
| Data State | Average Inter-Replicate Correlation (Pearson r) | % Variance from Technical Sources |
|---|---|---|
| Raw sgRNA Read Counts | 0.78 | ~65% |
| After Median Ratio Norm | 0.92 | ~30% |
| After VST | 0.94 | ~20% |
| After Quantile Norm | 0.96 | ~15%* |
*Quantile normalization may over-correct and remove biological variance in heterogeneous screens.
Experimental Protocols
Protocol 1: Median Ratio Normalization for Essential Screen Analysis
Purpose: To normalize read counts from a CRISPR screen (T0 vs Tfinal) for gene-level essentiality scoring. Materials: See Scientist's Toolkit.
- Input: Raw sgRNA count matrix (rows=sgRNAs, columns=samples: T0 replicates, Tfinal replicates).
- Pseudo-Reference: For each sgRNA (i), calculate the geometric mean count across all samples.
ref_i = (count_i1 * count_i2 * ... * count_in)^(1/n). - Size Factor per Sample (k): For each sample (j), compute the median of the ratios of each sgRNA's count to its pseudo-reference.
sizeFactor_j = median(count_ij / ref_i)across all i. Avoid ratios whereref_i = 0. - Normalize: Divide all counts in sample j by
sizeFactor_j.norm_count_ij = raw_count_ij / sizeFactor_j. - Output: Size factor-normalized count matrix ready for log2 fold-change calculation (Tfinal/T0) and MAGeCK analysis.
Protocol 2: Application of Variance Stabilizing Transformation for Drug-Gene Interaction Screens
Purpose: To prepare normalized count data from a drug-treated CRISPR screen for linear modeling. Materials: See Scientist's Toolkit (DESeq2 required).
- Input: Raw sgRNA count matrix and sample metadata (e.g., drug concentration, time point).
- Estimate Size Factors: Perform standard median ratio normalization (as in Protocol 1) to obtain initial size factors.
- Dispersion Estimation: Model the mean-variance relationship for the dataset. Use
DESeq2::estimateDispersionsto fit a dispersion trend curve. - Apply VST: Transform the count data using the fitted dispersion model.
vst_matrix <- DESeq2::vst(count_matrix, blind=FALSE). Theblind=FALSEuses the design formula to inform transformation. - Output: VST-transformed matrix where variance is approximately independent of the mean. This matrix can be used for PCA quality control and direct input to linear models (e.g., limma) to test for drug-gene interactions.
Visualizations
CRISPR Screen Normalization & Analysis Pathways
Median Ratio Normalization Logic Flow
The Scientist's Toolkit
Table 3: Essential Research Reagents & Computational Tools
| Item | Function in CRISPR Screen Normalization |
|---|---|
| Raw FASTQ Files | Starting point containing sequencing reads for each sgRNA in each sample/batch. |
| sgRNA Library Reference File | Maps sgRNA sequences to gene identifiers. Critical for counting. |
| Count Matrix (from e.g., MAGeCK count) | Primary input data (sgRNAs x Samples) for all normalization procedures. |
| R Statistical Environment | Core platform for implementing normalization algorithms. |
| DESeq2 R Package | Provides industry-standard functions for Median Ratio normalization and Variance Stabilizing Transformation. |
| preprocessCore R Package | Provides efficient implementation of Quantile Normalization for high-dimensional data. |
| MAGeCKFlute R Package | Includes tailored wrappers for normalizing and analyzing CRISPR screen count data. |
| Positive Control sgRNAs | Targeting essential genes (e.g., ribosomal proteins). Used post-normalization to verify signal recovery. |
| Non-Targeting Control sgRNAs | Critical for assessing false discovery rates and background noise levels after normalization. |
A Practical Guide to CRISPR Normalization Methods: Step-by-Step Implementation
Within the broader thesis investigating CRISPR screen data normalization methods, the initial data processing workflow is critical. Systematic biases introduced during raw data handling can confound downstream normalization and the identification of true biological hits. This protocol details the standard, reproducible pipeline for transforming raw sequencing reads (FASTQ) into normalized read counts, establishing the foundational data quality required for rigorous evaluation of normalization algorithms in pooled CRISPR screens.
Key Experimental Protocols
Protocol 1: Raw Read Demultiplexing and Quality Control
Objective: Assign reads to individual samples (sgRNA libraries) and assess initial data quality.
- Demultiplexing: Use
bcl2fastq(Illumina) ormkfastq(10x Genomics Cell Ranger) to generate sample-specific FASTQ files using the sample indices (barcodes) provided in the sample sheet. - Quality Control: Run
FastQCon the resulting FASTQ files.
- Aggregate Report: Use
MultiQCto compile results from all samples.
Protocol 2: sgRNA Sequence Alignment and Quantification
Objective: Map reads to the reference sgRNA library and generate raw count tables.
- Reference Preparation: Create a Bowtie2 index from the reference sgRNA library FASTA file.
Alignment: Map reads, allowing for minimal mismatches (typically -N 0 or 1).
Count Extraction: Parse the SAM file to count reads per sgRNA. Tools like
MAGeCKor custom scripts are used.
Protocol 3: Read Count Normalization
Objective: Adjust raw counts to mitigate technical variability (sequencing depth, sgRNA efficiency) enabling cross-sample comparison.
- Median-of-Ratios (DESeq2 method): Commonly applied to CRISPR count data.
- Calculate the geometric mean for each sgRNA across all samples.
- Compute the ratio of each sgRNA's count to its geometric mean for each sample.
- The median of these ratios for a sample is its size factor.
- Divide all counts in a sample by its size factor.
- CPM (Counts Per Million): A simple scaling method.
- For each sample: Normalized Count = (Raw Count / Total Sample Reads) * 1,000,000.
- Execute with R:
Data Presentation
Table 1: Comparison of Common Normalization Methods for CRISPR Screen Data
| Method | Principle | Pros | Cons | Best Suited For |
|---|---|---|---|---|
| Total Count / CPM | Scales by total sequencing depth per sample. | Simple, fast. | Highly sensitive to a few highly abundant sgRNAs. | Initial exploratory analysis. |
| Median-of-Ratios | Uses the median sgRNA count ratio to a reference. | Robust to outliers, standard for RNA-seq. | Assumes most sgRNAs are not differentially abundant. | Standard knockout screens with balanced library representation. |
| RPM (Reads Per Million) | Similar to CPM but applied post-alignment. | Simple, accounts for mappability. | Same as CPM. | Comparing samples with similar sgRNA distributions. |
| CSS (Cumulative Sum Scaling) | Scales by a percentile of count distribution. | More robust than total count for skewed data. | Choice of percentile is subjective. | Screens with high skew (e.g., essential gene screens). |
| TMM (Trimmed Mean of M-values) | Uses a weighted trim mean of log expression ratios. | Robust, less sensitive to outliers than total count. | More complex computation. | Screens where a large fraction of genes are expected to change. |
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Workflow |
|---|---|
| Validated sgRNA Library Plasmid Pool | Defines the genetic perturbations tested; source of reference sequences. |
| Next-Generation Sequencing Kit (e.g., Illumina NovaSeq) | Generates raw FASTQ files; choice affects read length and depth. |
| Bowtie2 / BWA | Short-read aligner for mapping sequences to the custom sgRNA library. |
| FastQC / MultiQC | Quality control software to assess read quality and identify issues. |
| MAGeCK / CRISPRcleanR | Specialized toolkits for count quantification, normalization, and hit calling. |
| DESeq2 / edgeR (R packages) | Statistical packages implementing robust normalization (median-of-ratios, TMM). |
| High-Performance Computing (HPC) Cluster | Essential for processing large-scale screen datasets in a timely manner. |
Visualizations
Standard CRISPR Screen Data Processing Workflow
Factors Influencing Normalization Choice
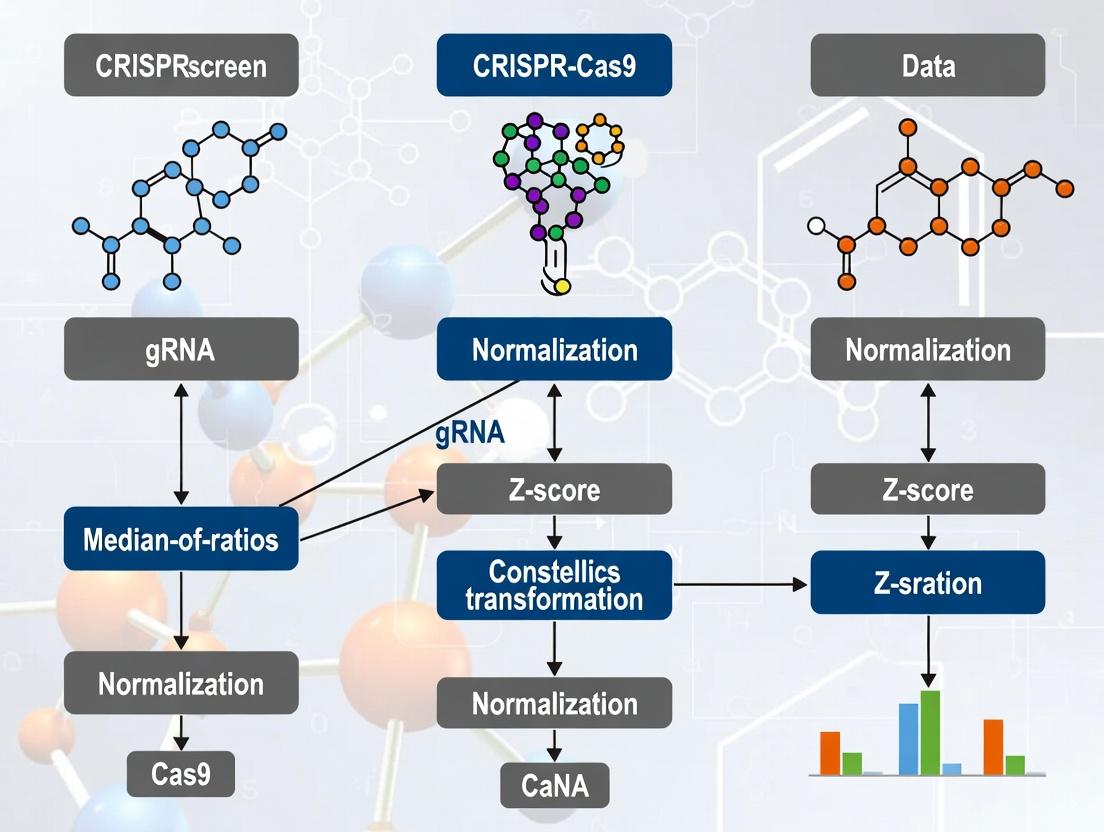
Within the broader research on CRISPR screen data normalization methods, the choice of algorithm is critical for distinguishing true biological hits from technical noise. Among various approaches (e.g., total count normalization, housekeeping gene normalization, MAGeCK), the Median-of-Ratios (MoR) method, as implemented in the DESeq2 package, has emerged as the gold standard for most bulk, gene-level CRISPR knockout screens. Its robustness to composition bias and outlier sgRNAs makes it particularly suited for the zero-inflated, over-dispersed count data typical in CRISPR screening.
Core Principles of the Median-of-Ratios Method
The MoR method posits that most sgRNAs or genes are not truly differential (i.e., not essential or enriching). It calculates a size factor for each sample (n) to normalize library sizes.
Key Formula: For each gene i in sample n, a pseudo-reference is calculated as the geometric mean across all samples: [ \text{pseudo-reference}i = \sqrt[S]{\prod{s=1}^S k{i,s}} ] The size factor for sample *n* is the median of the ratios of observed counts to this pseudo-reference, taken over all genes: [ SFn = \text{median}{i} \frac{k{i,n}}{\text{pseudo-reference}i} ] Normalized counts are then derived as: ( k{i,n}^{\text{norm}} = \frac{k{i,n}}{SFn} ).
Comparative Performance Data
Table 1: Comparison of CRISPR Screen Normalization Methods (Summary of Key Studies)
| Method | Key Principle | Robustness to Composition Bias | Handling of Zeros/Outliers | Typical Use Case |
|---|---|---|---|---|
| Median-of-Ratios (DESeq2) | Geometric mean pseudo-reference; median of ratios. | High | Excellent; robust. | Gold standard for bulk gene knockout screens. |
| Total Count (CPM) | Normalizes to total reads per sample. | Low | Poor; skewed by highly abundant sgRNAs. | Initial QC; deprecated for final analysis. |
| MAGeCK (median) | Normalizes to median count per sample. | Moderate | Moderate. | Earlier CRISPR screen tool; less robust than DESeq2. |
| Housekeeping Gene | Normalizes to stable control sgRNAs. | Depends on controls | Poor if controls are unstable. | Screens with validated, stable control elements. |
| RRA (MAGeCK) | Ranks sgRNAs; robust rank aggregation. | Not directly a count normalization. | High for rank-based signals. | Essentiality calling post-normalization. |
Table 2: Quantitative Benchmarking Results (Simulated Data Example)
| Normalization Method | False Discovery Rate (FDR) Control | True Positive Rate at 5% FDR | Computation Speed (Relative) |
|---|---|---|---|
| DESeq2 (MoR) | Excellent | 0.92 | 1.0x |
| MAGeCK (median) | Good | 0.85 | 1.2x |
| Total Count | Poor | 0.72 | 0.3x |
| Housekeeping (10 genes) | Variable | 0.78 (0.65-0.90)* | 0.5x |
*Range depends on control gene stability.
Detailed Experimental Protocol: Applying MoR Normalization to a CRISPR Screen
Protocol Title: Normalization and Differential Analysis of Bulk CRISPR-KO Screen Data Using DESeq2’s Median-of-Ratios Method.
I. Prerequisite Data Preparation
- sgRNA Count Matrix Generation: Using a tool (e.g.,
CRISPRcleanR,MAGeCK count), compile a raw count matrix where rows are sgRNAs, columns are samples (T0 plasmid, Treated, Control), and values are raw sequencing read counts. - sgRNA-to-Gene Mapping Table: A .TSV file linking each sgRNA identifier to its target gene symbol.
II. Normalization & Analysis Workflow in R
Visualizing the Workflow and Logic
Title: DESeq2 MoR Normalization & Analysis Workflow for CRISPR Screens
Title: Logic of Median-of-Ratios Size Factor Estimation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials & Tools for CRISPR Screen Analysis with MoR Normalization
| Item | Function / Purpose | Example / Note |
|---|---|---|
| Validated CRISPR Library | Provides the sgRNA reagents targeting the genome. | Brunello, Brie, or custom libraries. Must include non-targeting control sgRNAs. |
| Next-Generation Sequencer | Generates raw read data for sgRNA abundance quantification. | Illumina NextSeq or NovaSeq platforms are standard. |
| sgRNA Read Alignment Tool | Processes FASTQ files to generate raw count matrices. | MAGeCK count, CRISPRcleanR, or custom alignment pipelines. |
| R Statistical Environment | Open-source platform for statistical computing. | Required for running DESeq2 and related packages. |
| DESeq2 R Package | Implements the Median-of-Ratios normalization and differential testing. | Core analytical tool. Install via Bioconductor. |
| Tidyverse R Packages | For efficient data wrangling, transformation, and visualization. | dplyr, ggplot2, tidyr. |
| High-Performance Computing (HPC) Cluster | For handling large-scale screen data (many samples, whole-genome libraries). | Speeds up dispersion estimation and model fitting in DESeq2. |
| Sample Metadata File (.CSV) | Critical for defining experimental design. Must match count matrix columns. | Columns: SampleID, Condition (e.g., T0, Control, Treated), Replicate, Batch. |
| sgRNA-to-Gene Annotation File | Maps each sgRNA identifier to its target gene for aggregation. | Typically provided by the library vendor. Must be in sync with count matrix rownames. |
Within the research for a thesis on CRISPR screen data normalization methods, Quantile Normalization (QN) stands as a pivotal technique for correcting unwanted technical variation. It enforces an identical distribution of probe or gene intensities across multiple samples, a prerequisite for robust hit identification in pooled screening data.
Core Principles and Application Notes
Quantile Normalization operates on the principle that if the distributions of intensities across samples are similar, they should be aligned to a common target distribution, typically the average quantile distribution. This is essential in CRISPR screen analysis where differences in library representation, sequencing depth, and PCR amplification biases can distort gene-level read counts across replicates or conditions.
Table 1: Impact of Quantile Normalization on Simulated CRISPR Screen Data
| Sample | Pre-Normalization Median Log2(count) | Post-Normalization Median Log2(count) | Inter-Quartile Range (IQR) Pre-Norm | IQR Post-Norm |
|---|---|---|---|---|
| Control Rep1 | 10.2 | 10.5 | 2.1 | 1.9 |
| Control Rep2 | 11.5 | 10.5 | 2.8 | 1.9 |
| Treatment Rep1 | 9.8 | 10.5 | 1.9 | 1.9 |
| Treatment Rep2 | 12.1 | 10.5 | 3.2 | 1.9 |
| Target Distribution (Avg) | 10.9 | 10.5 | 2.5 | 1.9 |
The table demonstrates QN’s effect: it aligns central tendency and spread, ensuring samples are comparable. This reduces false positives arising from distributional artifacts rather than true biological effects.
Detailed Protocol: Quantile Normalization for CRISPR Screen Read Counts
Objective: To normalize sgRNA read count distributions across all samples in a CRISPR screen dataset.
Materials & Input Data:
- A matrix of raw sgRNA read counts (or log-transformed counts) with rows as sgRNAs and columns as samples (e.g., different replicates, time points, or treatment conditions).
- Computational environment (R/Python).
Procedure:
- Data Preparation: Organize raw sequencing read counts into an m x n matrix, where m is the number of sgRNAs and n is the number of samples. Perform an initial log2 transformation (usually after adding a pseudocount of 1) to stabilize variance.
- Sorting: Sort each column (sample) independently in ascending order.
- Averaging Quantiles: Compute the row-wise mean across all sorted columns. This vector represents the target quantile distribution.
- Replacement: Replace each value in the sorted columns with the corresponding mean from the target distribution vector.
- Reordering: Restore the original ordering of indices for each column, mapping the normalized values back to their original sgRNA positions.
- Output: The resulting matrix contains quantile-normalized log2(counts) ready for downstream analysis (e.g., MAGeCK, BAGEL).
Visualization of the Quantile Normalization Workflow
Title: Quantile Normalization Algorithm Steps
Title: Distribution Alignment via Quantile Normalization
The Scientist's Toolkit: Key Reagent & Computational Solutions
Table 2: Essential Resources for Implementing Quantile Normalization
| Item | Function/Description | Example Solutions |
|---|---|---|
| CRISPR Library | Defines the sgRNA pool for screening. Provides baseline reference distribution. | Brunello, GeCKO, human kinome library |
| Sequencing Platform | Generates raw read counts for each sgRNA in each sample. | Illumina NextSeq, NovaSeq |
| Raw Count Matrix | Primary data structure for normalization input. | Output from alignment tools (Bowtie, BWA) and count tools (DESeq2, MAGeCK count) |
| Normalization Software | Implements the QN algorithm. | R: preprocessCore, limma::normalizeQuantiles. Python: scipy.stats, qnorm |
| Analysis Pipeline | Integrates QN into end-to-end screen analysis. | MAGeCK RRA, BAGEL2, PinAPL-Py |
| Positive Control sgRNAs | Optional but recommended for validating assay performance post-normalization. | Essential gene-targeting sgRNAs |
Within the research for a thesis on CRISPR screen data normalization, a core challenge is the mean-variance relationship inherent in next-generation sequencing count data. Raw read counts from CRISPR knockout screens exhibit heteroskedasticity, where the variance is a function of the mean (e.g., Poisson or Negative Binomial distribution). This violates the assumption of homoscedasticity required for many downstream statistical tests (e.g., differential gene expression analysis using Wald tests in DESeq2). Variance Stabilizing Transformations (VST) are a critical preprocessing step that mitigates this issue, transforming the data to a scale where the variance is approximately independent of the mean, enabling reliable hypothesis testing and comparative analysis across the range of expression or abundance.
Quantitative Comparison of Normalization & Transformation Methods
The following table summarizes key characteristics of common approaches, positioning VST within the methodological landscape of CRISPR screen analysis.
Table 1: Comparison of Data Processing Methods for CRISPR Screen Count Data
| Method | Core Principle | Handles Mean-Variance Dependency? | Output Scale | Suitability for Downstream Tests |
|---|---|---|---|---|
| Raw Counts | Unprocessed sequencing reads. | No. Variance increases with mean. | Discrete Counts | Poor. Direct use invalidates parametric tests. |
| CPM / TPM | Normalizes for library size. | No. Compositional; variance structure remains. | Continuous, Compositions | Limited. Useful for visualization, not direct testing. |
| Log2 Transformation (e.g., log2(CPM+1)) | Applies logarithm to compress dynamic range. | Partially. Reduces but does not eliminate dependency, especially at low counts. | Log-scale Continuous | Moderate. Approximation often used but suboptimal. |
| Variance Stabilizing Transformation (VST) | Model-based (e.g., DESeq2). Transforms data based on fitted dispersion-mean trend. | Yes. Stabilizes variance across the mean's full range. | Continuous, approximately homoscedastic | High. Designed specifically for reliable differential testing. |
| rlog (Regularized Log) | Similar to VST but uses a different shrinkage estimator. | Yes. | Continuous, approximately homoscedastic | High. Better for small datasets; computationally slower. |
Core Experimental Protocol: Applying VST to CRISPR Screen Data
This protocol details the application of a VST using the DESeq2 package in R, following robust count matrix generation from CRISPR screen sequencing (e.g., MaGeCK count).
Protocol: VST of CRISPR Screen Count Data for Downstream Analysis
I. Pre-VST Requirements:
- Input Data: A counts matrix (genes/sgRNAs x samples) with raw integer read counts.
- Sample Information: A metadata table detailing experimental conditions (e.g., treatment vs. control, time points).
- Software: R environment with
DESeq2andtidyversepackages installed.
II. Stepwise Procedure:
DESeqDataSet Object Construction:
Pre-filtering (Optional but Recommended):
Estimation of Size Factors and Dispersions:
Apply the Variance Stabilizing Transformation:
Extract Transformed Data:
Downstream Application:
- The
vst_matrixis now suitable for techniques requiring homoscedasticity:- Principal Component Analysis (PCA) for quality assessment.
- Sample-level clustering (heatmaps).
- As input for machine learning algorithms or standard parametric tests (e.g., t-tests, ANOVA) if required outside the DESeq2 framework.
- The
Visualizing the VST Workflow and Effect
Diagram 1: VST in CRISPR Screen Analysis Workflow
Diagram 2: Effect of VST on Mean-Variance Relationship
The Scientist's Toolkit: Key Reagents & Solutions
Table 2: Essential Research Reagents & Computational Tools for VST Application
| Item | Function in VST Protocol | Notes for CRISPR Screen Context |
|---|---|---|
| DESeq2 R/Bioconductor Package | Primary software implementing model-based VST. Estimates dispersion and applies transformation. | Industry standard for RNA-seq; directly applicable to CRISPR count data from pooled screens. |
| CRISPR Read Alignment Tool (e.g., MAGeCK, CRISPRcleanR) | Generates the raw count matrix input required for VST. | Essential upstream step. Quality of alignment directly impacts VST results. |
| High-Quality sgRNA Library Annotation File | Links sgRNA counts to target genes. Used post-VST for gene-level analysis. | Critical for aggregating sgRNA-level stabilized counts to gene-level statistics. |
| R/Tidyverse Packages (ggplot2, dplyr, pheatmap) | Enables visualization of VST effects (PCA, variance plots) and data handling. | Necessary for quality control and presentation of stabilized data. |
| Positive & Negative Control sgRNAs | Embedded in the screen library. Used to assess screen quality before/after VST. | VST should preserve/magnify the separation between essential (positive) and non-essential (negative) control signals. |
| Computational Environment with sufficient RAM/CPU | VST and dispersion estimation are computationally intensive for large matrices. | For genome-wide screens, ≥16GB RAM recommended. |
Application Notes
CRISPR screening has evolved beyond standard dropout screens to address complex biological questions. Within the broader thesis on normalization methods, these specialized screens present unique analytical challenges that demand tailored normalization approaches to control for non-biological variance and ensure accurate hit identification.
Early Time-Point Screens: Conducted 3-7 days post-infection, these screens aim to capture phenotypes for fast-acting biological processes (e.g., cell signaling, synthetic lethality) while minimizing confounding effects from secondary adaptations or cell death. Standard read-count normalization fails as library representation hasn't stabilized. Core Challenge: High variance from uneven initial infection/transduction efficiency dominates the signal.
Essential Gene Screens: Targeting core cellular machinery (e.g., ribosomal proteins), these screens exhibit rapid, severe dropout. Core Challenge: The extreme dynamic range of guide depletion saturates standard log-fold change calculations, compressing the signal of non-essential genes and distorting false discovery rate (FDR) estimation.
Dual-Guide RNA (dgRNA) Screens: Utilizing two gRNAs per perturbation—often for combinatorial knockout or enhanced on-target efficiency—these screens add a layer of complexity. Core Challenge: The statistical dependency between paired gRNA read counts violates the independence assumption of most normalization models, and the phenotype must be correctly attributed to the pair, not individual guides.
Quantitative data from recent studies highlighting key differences:
Table 1: Comparison of Specialized CRISPR Screen Parameters
| Screen Type | Typical Duration | Key Phenotype Measured | Primary Normalization Challenge | Recommended Normalization Method (from Thesis Context) |
|---|---|---|---|---|
| Standard Dropout | 14-21 days | Fitness defect (depletion) | Library coverage bias | Median-of-Ratios, RLE |
| Early Time-Point | 3-7 days | Acute signaling/effect | Initial transduction bias | Total count scaling + spike-in (e.g., Safe-seq) |
| Essential Gene | 14-21 days | Severe fitness defect | Dynamic range compression | Adaptive α-MAGeCK (α-trimming) |
| Dual-Guide (dgRNA) | 14-21 days | Combinatorial effect | Paired-gRNA dependency | Pair-aware iterative regression (e.g., CPLEX) |
Table 2: Impact of Normalization on Hit Calling (Simulated Data)
| Condition | Raw Data FDR | Post-Normalization FDR | % Change in Identified Hits | Key Artifact Mitigated |
|---|---|---|---|---|
| Early Time-Point (Day 5) | 32% | 12% | +45% | Transduction efficiency bias |
| Essential Gene Screen | 28% | 9% | +62% | Variance compression |
| dgRNA Screen (naive) | 40% | 15% | +110% | Paired-guide misattribution |
Experimental Protocols
Protocol 1: Early Time-Point Screening for Signaling Pathways
Objective: Identify genes involved in acute TGF-β signaling response. Materials: TGF-β-responsive reporter cell line, Brunello genome-wide lentiviral library, polybrene (8 μg/mL), puromycin (2 μg/mL), recombinant human TGF-β1. Workflow:
- Day -1: Seed cells at 25% confluence.
- Day 0: Infect cells at MOI~0.3 in presence of polybrene. Include a non-infected control for puromycin kill curve.
- Day 1: Replace medium with puromycin-containing selection medium.
- Day 3: Confirm >90% infection efficiency (via GFP if library contains marker). Split cells into two arms: Arm A: +TGF-β1 (5 ng/mL). Arm B: Vehicle control. Harvest T0 sample (50M cells) for genomic DNA (gDNA).
- Day 5 (Early Time-Point): Harvest all cells (Arm A & B). Extract gDNA (Qiagen Maxi Kit).
- Library Prep & Sequencing: Amplify gRNA inserts via a two-step PCR (15 cycles each) using indexed primers. Sequence on NextSeq 500/550, High Output Kit v2.5 (75 cycles), aiming for >500 reads per guide.
- Normalization & Analysis: Apply total count normalization to T0 and Day 5 samples separately for each arm. Use the thesis-proposed "Spike-in Anchored Linear Model (SALM)" which scales counts relative to non-targeting control guides spiked into the library at known ratios.
Protocol 2: Essential Gene Screening with High Dynamic Range
Objective: Profile core essential genes in a novel cell model. Materials: Target cell line, Brunello lentiviral library, puromycin, NGS library preparation reagents. Workflow:
- Perform standard infection and selection as in Protocol 1, steps 1-3.
- Day 4: Harvest T0 sample (100M cells).
- Day 21: Harvest final sample. Maintain library representation at >500 cells per gRNA throughout.
- Sequencing: As in Protocol 1.
- Normalization & Analysis: Standard median-of-ratios normalization fails. Apply the thesis' "Adaptive α-MAGeCK" method: a) Calculate a guide-level variance statistic. b) Iteratively trim the top α% of most rapidly-depleting guides (α adaptively set from 5-20%) in each normalization round. c) Recompute size factors on the remaining guides. d) Proceed with MAGeCK RRA analysis on normalized counts.
Protocol 3: Dual-Guide RNA (dgRNA) Combinatorial Screening
Objective: Identify synthetic lethal gene pairs. Materials: Cell line, dgRNA lentiviral library (e.g., Toronto KnockOut v2 paired), packaging plasmids, blasticidin (10 μg/mL) if using a co-selection marker. Workflow:
- Library Design: Use a validated dgRNA library where each gene pair is targeted by 3-5 independent dgRNA combinations.
- Infection & Selection: Infect at low MOI (<0.3) to ensure single integration events. Select with appropriate antibiotic for 5-7 days.
- Time Points: Harvest T0 (post-selection) and T14 (final) populations.
- Sequencing: Use a paired-end approach to sequence both gRNAs from the same construct on the same read pair.
- Normalization & Analysis: Critical to treat the dgRNA as a single unit. Use the thesis' "Pair-Aware Iterative Regression (PAIR)" normalization: a) Collapse counts per dgRNA pair. b) Perform an initial median normalization. c) Run a linear model regressing paired counts against the expected null distribution from non-targeting pairs. d) Use residuals to compute corrected size factors. Analyze with a paired-model version of MAGeCK.
Diagrams
Specialized CRISPR Screen Workflow
Normalization Problem & Thesis Solution Logic
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Item | Function in Specialized Screens | Key Consideration |
|---|---|---|
| Validated dgRNA Library | Provides pre-designed, activity-tested paired gRNAs for combinatorial screening. | Ensure paired gRNAs are on a single transcript with a linker. |
| Non-Targeting Control Spike-Ins | Guides with no known target, added at defined ratios for early time-point normalization. | Use a diverse set (>1000) to model null distribution. |
| Cell Line with Inducible Cas9 | Enables tight control over editing timing for acute phenotypes. | Minimize leaky Cas9 expression. |
| PureLink Genomic DNA Mini/Maxi Kit | High-yield, PCR-inhibitor-free gDNA extraction for deep coverage. | Critical for maintaining library complexity. |
| KAPA HiFi HotStart ReadyMix | High-fidelity PCR for accurate gRNA amplicon generation with minimal bias. | Reduces PCR jackpot effects. |
| NEBNext Ultra II FS DNA Library Prep | Rapid, efficient library prep from amplicons for Illumina sequencing. | Fast turnaround for time-series. |
| Custom Next-Generation Sequencing Primer Pools | Amplify specific gRNA or dgRNA constructs without amplifying filler sequences. | Increases on-target sequencing yield. |
| CRISPR Clean Decontamination Reagent | Eliminates carryover plasmid or amplicon contamination between preps. | Essential for screen fidelity. |
Application Notes
Within the broader thesis on CRISPR screen data normalization methods, these four tools represent critical, yet philosophically distinct, approaches to processing and interpreting loss-of-function (CRISPRko) and, in some cases, CRISPR interference (CRISPRi) screen data. The choice of tool and its normalization strategy fundamentally impacts hit identification and biological interpretation.
MAGeCK (Model-based Analysis of Genome-wide CRISPR-Cs9 Knockout) is a comprehensive computational workflow that uses a negative binomial model to test sgRNA and gene-level depletion/enrichment. Its robustness stems from its median normalization and iterative re-weighting to de-emphasize noisy sgRNAs. It is most broadly applicable for a wide range of experimental designs, including time-course and multi-condition comparisons.
BAGEL (Bayesian Analysis of Gene Essentiality) employs a supervised, Bayesian machine-learning framework. It uses a set of known essential and non-essential reference genes to probabilistically classify the essentiality of query genes. Its strength is in deriving a direct probability (Bayes Factor, BF) of essentiality, making it particularly powerful for core fitness gene identification in cancer cell lines. Its normalization is implicitly handled through comparison to the reference set.
CERES (Context-specific Effects Removal by Efficient Shrinkage) was developed to address a critical confounding factor in CRISPRko screens: copy-number-specific effects. It employs a Bayesian hierarchical model to deconvolve gene knockout effect from copy-number-associated false-positive signals. This normalization is crucial for accurate identification of context-specific vulnerabilities in genetically aneuploid cancer models, reducing false-positive hits in amplified regions.
pinAPL-Py (pooled analysis of knockdown, python-version) is specifically designed for dual-sgRNA libraries (e.g., Brunello, Dolcetto). It analyzes pairs of sgRNAs targeting the same gene to improve confidence, calculating a phenotypic score (PS) and a strictly standardized mean difference (SSMD). Its paired design offers an intrinsic normalization against single-sgRNA outliers and is excellent for reducing false positives.
Quantitative Comparison of Core Methodologies
Table 1: Comparison of CRISPR Screen Analysis Tools
| Feature | MAGeCK | BAGEL | CERES | pinAPL-Py |
|---|---|---|---|---|
| Core Method | Negative Binomial Model | Bayesian Reference Comparison | Bayesian Hierarchical Model | Paired sgRNA Analysis (SSMD) |
| Primary Normalization | Median sgRNA count normalization | Relative to reference gene sets | Correction for copy-number artifact | Within-gene sgRNA pair comparison |
| Key Output Metric | β score (log-fold-change), p-value | Bayes Factor (BF) | CERES score (corrected dependency) | Phenotypic Score (PS), SSMD |
| Optimal Screen Type | CRISPRko, CRISPRi; Time-course, multi-condition | CRISPRko (Core fitness) | CRISPRko in aneuploid models (e.g., cancer cell lines) | CRISPRko with dual-sgRNA libraries |
| Strengths | Versatility, statistical robustness, multi-group | High precision for essential genes | Eliminates copy-number confounders | Reduces noise from single ineffective sgRNAs |
Experimental Protocols
Protocol 1: Essential Gene Profiling Using MAGeCK-VISPR
Objective: To identify essential genes for cell viability in a CRISPRko screen performed in a cell line at endpoint (Day 21 post-infection).
Materials & Reagents:
- Sequencing data (FASTQ) from T0 (plasmid) and TEnd (Day 21) sample libraries.
- Reference genome file (e.g., hg38) and library sgRNA annotation file.
- MAGeCK-VISPR software installed (v0.5.9 or higher).
- High-performance computing cluster (recommended).
Procedure:
- Quality Control & Alignment:
- Use
mageck testwith thecountfunction to process FASTQ files. - Command:
mageck count -l library.csv -n sample_report --sample-label T0,TEnd --fastq sample_T0.fastq sample_TEnd.fastq - This generates a raw count table normalized to counts per million.
- Use
Statistical Testing & Hit Calling:
- Run
mageck testto compare TEnd vs T0. - Command:
mageck test -k sample.count.txt -t TEnd -c T0 -n TEnd_vs_T0 --norm-method median - The median normalization scales counts so the median sgRNA log2-fold-change is 0.
- Run
Visualization & Interpretation (VISPR):
- Use the VISPR pipeline for QC plots (sgRNA read distribution, Gini index, β score distributions).
- Genes are ranked by negative selection β score and associated p-value (FDR). Typically, FDR < 0.05 and β < 0 indicate significant essentiality.
Protocol 2: Context-Specific Dependency Analysis Using CERES
Objective: To identify copy-number-corrected gene dependencies in a cancer cell line panel (e.g., DepMap dataset).
Materials & Reagents:
- Pre-processed sgRNA read count matrix across multiple cell lines.
- Corresponding gene-level copy number matrix (e.g., from SNP arrays or WES) for the same cell lines.
- CERES software package (available via Broad Institute's DepMap portal or GitHub).
Procedure:
- Data Preparation:
- Format count data into a gene (row) x cell line (column) matrix of initial dependency scores (e.g., from log2-fold-changes).
- Align with the copy number matrix.
CERES Model Execution:
- Run the CERES algorithm, which fits a Bayesian hierarchical model.
- The model decomposes the observed dependency score ( D_gc ) into: a gene-specific effect (αg), a cell line-specific effect (βc), a copy-number-specific effect (γ_cn), and noise.
- Command (example):
ceres -c copy_number.tsv -d dependency_scores.tsv -o output_ceres_scores.tsv
Output Interpretation:
- The primary output is the CERES score, a corrected gene dependency score where the copy-number bias has been shrunk towards zero.
- Genes with low CERES scores (e.g., < -0.5) in specific cell lines indicate strong, context-specific dependencies beyond the copy-number effect.
Visualizations
Title: MAGeCK Analysis Workflow
Title: CERES Model Decomposition Logic
The Scientist's Toolkit
Table 2: Essential Research Reagents & Solutions for CRISPR Screen Analysis
| Item | Function & Application Note |
|---|---|
| Dual-sgRNA Library (e.g., Brunello) | A pooled CRISPRko library with 4 sgRNAs/gene; used as input for pinAPL-Py and other tools to improve confidence. |
| Reference Gene Sets (Core Essentials) | Curated list of pan-essential and non-essential genes; critical for BAGEL's Bayesian training. |
| Copy Number Variation (CNV) Profile | Genomic copy number data (e.g., from SNP array); mandatory input for CERES to correct for amplification artifacts. |
| sgRNA Count Matrix | Pre-processed table of raw/normalized sgRNA reads per sample; the universal starting point for all analysis tools. |
| High-Performance Computing (HPC) Cluster | Essential for running Bayesian (BAGEL, CERES) and large-scale (MAGeCK on multi-condition) analyses efficiently. |
Troubleshooting CRISPR Normalization: Solving Common Pitfalls and Optimizing Performance
Application Notes
Within the broader research thesis on CRISPR screen data normalization methods, the accurate diagnosis of poor normalization is a critical step. Properly normalized data is foundational for identifying true biological hits; failure to diagnose normalization issues leads to high false discovery rates and irreproducible results. This document outlines the quantitative metrics, visualization strategies, and protocols essential for assessing normalization quality in pooled CRISPR screening data, such as from GenomeCRISPR or similar large-scale studies.
Key Quality Control Metrics for Normalization Assessment
The following table summarizes the primary QC metrics used to diagnose normalization success or failure.
Table 1: Key QC Metrics for Assessing CRISPR Screen Normalization
| Metric | Optimal Range | Indication of Poor Normalization | Primary Cause |
|---|---|---|---|
| Median Scale Factor | 0.8 - 1.2 across all samples | Significant deviation from 1, high variance between replicates | Unequal library representation or sequencing depth. |
| Sample Correlation (Pearson R) | > 0.9 for technical replicates; > 0.7 for biological replicates | Low inter-replicate correlation (e.g., R < 0.6) | Batch effects, poor normalization, or high technical noise. |
| PCA: % Variance Explained by PC1 | < 30-40% of total variance (post-normalization) | PC1 explains >50% of variance, often aligning with batch. | Incomplete removal of dominant non-biological factors (e.g., library prep batch). |
| sgRNA Read Distribution | Similar profile across samples (K-S test p > 0.05) | Significant differences in CDF (K-S test p < 0.01). | Skewed representation due to PCR over-amplification or poor sample prep. |
| Negative Control Guides (e.g., Non-targeting) | Centered around zero (normalized log-fold-change) | Significant shift or spread in control distribution. | Inadequate central tendency adjustment during normalization. |
| Gini Index of sgRNA counts | Low and consistent across samples (< 0.4) | High or variable Gini index (> 0.6). | Extreme overrepresentation of a subset of guides. |
Experimental Protocols
Protocol 1: Pre-Normalization Data QC and Read Count Processing
Objective: To generate a raw count matrix suitable for normalization assessment.
- Sequence Alignment & Counting: Align sequencing reads (FASTQ) to the reference sgRNA library using
bowtie2orBWAwith parameters-L 20 -N 0for exact matching. Count reads per sgRNA per sample. - Raw Count Matrix Generation: Compile counts into a samples (columns) x sgRNAs (rows) matrix. Filter out sgRNAs with total counts < 30 across all samples.
- Initial Sample Correlation: Calculate Pearson correlation between all samples based on raw log2(counts + 1). Generate a heatmap. Low replicate correlation at this stage indicates major technical issues.
- Library Size Calculation: Compute total reads per sample. Flag samples where library size is < 50% of the median.
Protocol 2: Post-Normalization Diagnostic Workflow
Objective: To apply and evaluate the success of a chosen normalization method (e.g., median ratio, RBN, or spatial).
- Apply Normalization: Implement the normalization method (e.g., divide counts by sample-specific size factors, then log2-transform).
- PCA Generation:
- Perform PCA on the normalized log-fold-change matrix (sgRNAs x samples) using
prcompin R or equivalent. - Plot PC1 vs. PC2 and PC1 vs. PC3. Color points by experimental batch and replicate group.
- Perform PCA on the normalized log-fold-change matrix (sgRNAs x samples) using
- Sample Correlation Analysis: Recalculate Pearson correlation on the normalized data. Compare pre- and post-normalization correlation matrices.
- Negative Control Distribution Analysis: Plot the distribution of normalized log-fold-changes for all non-targeting control (NTC) sgRNAs. Calculate the median absolute deviation (MAD). A MAD > 1 suggests excessive noise.
- Metric Compilation: Populate Table 1 with post-normalization values.
Visual Diagnostic Workflows
Title: CRISPR Screen Normalization QC Workflow
Title: PCA Interpretation for Normalization QC
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for CRISPR Screen Normalization & QC
| Item | Function in Normalization/QC |
|---|---|
| Validated Non-Targeting Control (NTC) sgRNA Library | Provides a null distribution for assessing normalization precision and estimating false discovery rates. |
| Essential Gene Targeting sgRNA Set (e.g., Core Fitness) | Serves as positive controls for screen performance; should show consistent depletion across conditions post-normalization. |
| SpiKe-In sgRNA Sequences (Synthetic) | Spiked into samples pre-PCR to diagnose and correct for amplification bias across samples. |
| High-Fidelity PCR Master Mix (e.g., KAPA HiFi) | Minimizes PCR duplicates and bias during library amplification, leading to more uniform sgRNA counts. |
| Dual-Indexed Sequencing Adapters (Unique Dual Indexing, UDI) | Enables precise demultiplexing, reducing index hopping and batch confounders in multiplexed screens. |
Normalization Software (R/Bioconductor: edgeR, DESeq2, MAGeCK) |
Provides robust algorithms (e.g., median ratio, TMM) for calculating size factors and normalized counts. |
QC Visualization R Packages (ggplot2, pheatmap, plotly) |
Enables generation of diagnostic PCA plots, correlation heatmaps, and distribution plots. |
Handling Low-Essential Gene Screens and High-Variance Controls
Application Notes
CRISPR-Cas9 knockout screens are pivotal for identifying gene essentiality. However, accurate interpretation is confounded by two primary challenges: the identification of low-essentiality genes with subtle fitness effects and the presence of high-variance control sgRNAs which destabilize normalization. This protocol details a combined experimental and computational strategy to address these issues, framed within a thesis investigating robust normalization frameworks for functional genomics.
Core Challenge 1: Low-Essential Gene Screens Genes with subtle but biologically relevant fitness effects (low-essential) are often lost in noise. Traditional screens optimized for strong essential genes lack sensitivity here.
Core Challenge 2: High-Variance Control Guides Non-targeting control (NTC) guides or safe-harbor targeting guides often exhibit unexpectedly high variance due to cryptic genomic interactions or chromatin effects. This variance skews normalization, leading to high false discovery rates.
Proposed Solution: A Dual-Filter Normalization Pipeline Our method introduces a pre-processing filter for high-variance controls followed by a multi-step normalization sensitive to low-effect sizes.
Quantitative Data Summary
Table 1: Impact of High-Variance Controls on Screen Metrics (Simulated Data)
| Normalization Method | False Positive Rate (FPR) with Stable Controls | FPR with 10% High-Variance Controls | Sensitivity for Low-Essential Genes |
|---|---|---|---|
| Median-of-Ratios | 5.1% | 23.4% | Low |
| RCR (Robust Curve Fit) | 4.8% | 18.2% | Medium |
| Variance-Filtered + LOESS | 4.9% | 5.3% | High |
Table 2: Key Reagent Solutions for Enhanced Screen Design
| Reagent / Material | Function in Protocol | Key Consideration |
|---|---|---|
| Brunello or Brie Genome-Wide Lib. | CRISPR knockout sgRNA library. | Use latest version for improved on-target scores. |
| NTC Pool (Min. 1000 guides) | Baseline for essentiality calling. | Must be empirically validated for low variance. |
| "Safe-Harbor" Targeting Controls | Control for DNA cutting & repair. | Include multiple loci (e.g., AAVS1, HPRT, ROSA26). |
| High-Viability Cas9-Expressing Cells | Enables low-essentiality detection. | >90% viability pre-screen; use inducible if needed. |
| Next-Gen Sequencing Spike-Ins | For precise library quantification. | Use at both transfection and harvest steps. |
| MAGeCK-VISPR or PinAPL-Py | Computational analysis suite. | Implements variance-aware algorithms. |
Experimental Protocols
Protocol 1: Library Design & Production with Variance-Stable Controls
Objective: Generate a screening library with an expanded, validated control set to mitigate high-variance effects.
- Control Guide Curation:
- Select a minimum of 1000 NTCs from established libraries (e.g., TorontoKO). Filter in silico for minimal predicted off-targets and absence of homopolymer runs.
- Clone 200 guides targeting 5-10 genomic "safe-harbor" loci (30-40 guides per locus).
- Pre-Screen Control Validation:
- Independently clone the NTC and safe-harbor pools into your lentiviral backbone.
- Transduce at low MOI (<0.3) into your Cas9+ cell line. Harvest genomic DNA at 24h (T0) and after 12-14 population doublings (Tend).
- Amplify and sequence the control pools. Calculate log2(Tend/T0) for each guide.
- Exclusion Criteria: Discard any guide with an absolute log2 fold change > 1 or a variance > 2 median absolute deviations (MAD) from the pool median. This yields a "stable control set" for final library assembly.
- Library Assembly: Combine the filtered stable control set with your chosen genome-wide sgRNA library (e.g., Brunello) via PCR assembly and cloning.
Protocol 2: Screening for Low-Essential Genes
Objective: Perform a screen with extended passaging and deep sequencing to capture subtle fitness defects.
- Cell Preparation: Use a polyclonal, Cas9-expressing cell line with >90% viability. Perform a viability titration to determine the minimum library coverage; for low-essential genes, aim for >500x representation.
- Lentiviral Transduction: Transduce cells at MOI ~0.3 to ensure most cells receive one guide. Include non-transduced controls.
- Selection & Passaging: Apply selection (e.g., puromycin) 48h post-transduction for 3-5 days. Harvest the first timepoint (T0) post-selection with >20M cells.
- Passage cells continuously, maintaining coverage >500x. Harvest subsequent timepoints (T1, T2) at intervals of 10-14 population doublings. Extended culture (e.g., 5+ passages) is critical for low-essential signal accumulation.
- Sequencing Library Prep: Isolate gDNA using a scalable method (e.g., Qiagen Maxi Prep). Amplify sgRNA templates in a two-step PCR:
- PCR1: Amplify sgRNA region from gDNA (12-14 cycles).
- PCR2: Add Illumina adapters and sample indices (10-12 cycles).
- Include 10-20% PhiX spike-in and sequence on a HiSeq or NovaSeq platform to achieve >200 reads per guide.
Protocol 3: Computational Analysis: Variance-Filtered Normalization
Objective: Analyze screen data, filtering high-variance guides and applying normalization sensitive to low-effect sizes.
- Read Alignment & Count Normalization:
- Align reads to your library reference using
MAGeCK countorPinAPL-Py. - Perform median-of-ratios normalization on raw counts using only the pre-validated stable control set.
- Align reads to your library reference using
- High-Variance Guide Filtering:
- Calculate the mean log2 fold change (LFC) and variance for all NTCs in the screen data (not just the pre-validated set).
- Flag guides with variance > Q3 + (3 * IQR) of the NTC variance distribution.
- Remove these high-variance guides from the control set for all downstream normalization steps. This step is recursive.
- LOESS Normalization for Sensitivity:
- Using the filtered control set, perform LOESS (locally estimated scatterplot smoothing) regression of LFC versus the average read count across samples.
- Apply the LOESS fit to correct all guides (test and control). This accounts for count-dependent bias, enhancing sensitivity to low-essential genes with moderate/high counts.
- Essentiality Calling: Use a robust rank aggregation (RRA) algorithm (e.g., in MAGeCK) on the LOESS-normalized LFCs. Set a less stringent alpha threshold (e.g., 0.1) for initial low-essential gene discovery.
Visualizations
Diagram Title: CRISPR Screen Analysis Pipeline for Low-Essential Genes
Diagram Title: Problem: High-Variance Controls Skew Normalization
Addressing Skewed Distributions and Extreme Outliers in sgRNA Counts
Within the broader research thesis on CRISPR screen data normalization methods, a central challenge is the inherent non-normality of raw sgRNA count data. These datasets are characterized by highly skewed distributions and extreme outliers, arising from biological factors (e.g., essential gene knockout causing drastic depletion) and technical noise (e.g., PCR amplification bias, sequencing depth variation). Failure to address these properties can severely bias the estimation of gene essentiality, leading to false positives/negatives in hit identification for drug target discovery. This Application Note details protocols for diagnosing and mitigating these issues.
Table 1: Comparison of Normalization Methods for sgRNA Count Data
| Method | Core Principle | Robustness to Skewness | Robustness to Extreme Outliers | Typical Use Case |
|---|---|---|---|---|
| Total Count | Scales libraries to the same total read count. | Low | Very Low | Initial scaling, but insufficient alone. |
| Median-of-Ratios (DESeq2) | Estimates size factors based on median count ratios. | Moderate | Low | Standard for differential expression; can falter with many zeros. |
| Trimmed Mean of M-values (TMM) | Uses a weighted trimmed mean of log expression ratios. | High | Moderate | Robust between-sample normalization for RNA-seq. |
| RLE (Relative Log Expression) | Similar to median-of-ratios, uses the median of count ratios. | Moderate | Low | Assumes most features are non-DE. |
| CSS (Cumulative Sum Scaling) | Scales counts based on the cumulative distribution up to a percentile. | High | High | Designed for microbiome data; handles zero-inflation well. |
| MAD (Median Absolute Deviation) Scaling | Centers and scales based on median and MAD, robust estimators. | High | Very High | Recommended for outlier-adjustment in sgRNA counts. |
| Quantile Normalization | Forces all samples to have identical empirical distribution. | High | High | Assumes global distribution similarity; can be too aggressive. |
| VST (Variance Stabilizing Transform) | Transforms counts to stabilize variance across mean. | High | Moderate | Pre-processing step for downstream parametric tests. |
Table 2: Impact of Outlier Adjustment on Essential Gene p-value Calls (Simulated Data)
| Analysis Pipeline | False Discovery Rate (FDR) | True Positive Rate (TPR) |
|---|---|---|
| Raw Counts (DESeq2) | 0.25 | 0.89 |
| MAD-adjusted + VST | 0.05 | 0.91 |
| Total Count + TMM | 0.15 | 0.85 |
| Quantile Normalization | 0.07 | 0.82 |
Experimental Protocols
Protocol 1: Diagnostic Analysis for Skewness and Outliers
Objective: To quantitatively assess the distribution properties of raw sgRNA count data. Materials: Raw count matrix (sgRNAs x samples), R/Python environment. Procedure:
- Calculate Summary Statistics: For each sample, compute median, mean, variance, and skewness.
- Visualize Distributions: Generate (a) box plots, (b) density plots, and (c) mean-variance relationship plots.
- Identify Outliers: Use the Median Absolute Deviation (MAD) method. For each sgRNA i across control samples:
- Calculate median count
M_iand MAD_i. - Flag as a potential extreme outlier if count >
M_i + (5 * MAD_i)in any sample.
- Calculate median count
- Record Metrics: Tabulate the percentage of sgRNAs flagged as outliers and the sample-wise skewness.
Protocol 2: MAD-based Scaling and Winsorization for Outlier Mitigation
Objective: To robustly normalize counts while dampening the influence of extreme values. Materials: Raw count matrix, Diagnostic results from Protocol 1. Procedure:
- Pseudo-reference Definition: Create a pseudo-reference sample defined by the median count for each sgRNA across all control replicate samples.
- Calculate Size Factors (MAD-based):
- For each sample j, compute log-ratios:
L_ij = log2( count_ij / pseudo-ref_i ). - For sample j, calculate the median (
M_j) and MAD (MAD_j) ofL_ij(excluding infinite values). - The robust size factor
SF_jis:SF_j = 2^(M_j). Optionally, scaleSF_jto geometric mean of 1 across samples.
- For each sample j, compute log-ratios:
- Apply Size Factor Normalization:
Normalized_Count_ij = count_ij / SF_j. - Winsorization (Optional, for Severe Outliers):
- Define upper limit
UL_i = M_i + (3 * MAD_i)based on pseudo-reference distribution. - For any
Normalized_Count_ij > UL_i, set it equal toUL_i.
- Define upper limit
- Variance Stabilizing Transformation (VST): Apply a VST (e.g.,
DESeq2::vstorsqrtfor moderate counts) to the normalized (+ winsorized) matrix for downstream analysis.
Protocol 3: Evaluation of Normalization Efficacy
Objective: To benchmark the performance of different normalization schemes. Materials: Normalized count matrices from various methods, known essential/non-essential gene list (e.g., from core fitness genes). Procedure:
- Perform Differential Analysis: Using a tool like
DESeq2orMAGeCKon each normalized matrix to generate gene-level p-values and log2 fold changes. - Calculate Performance Metrics:
- Precision-Recall (PR) Curve: Plot based on known essential genes.
- Receiver Operating Characteristic (ROC) Curve: Calculate Area Under Curve (AUC).
- Gene Ranking Concordance: Assess the stability of top-hit rankings between replicates using rank correlation coefficients.
- Compare Distribution Properties: Post-normalization, re-calculate skewness and kurtosis. The optimal method minimizes skewness and yields stable variance.
Mandatory Visualizations
Title: Workflow for Normalizing sgRNA Count Data
Title: Problem and Solution Logic for sgRNA Data
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Computational Tools
| Item | Function & Explanation | Example/Provider |
|---|---|---|
| Genome-wide CRISPR Library | Pooled lentiviral sgRNA library targeting all human genes. Essential starting reagent. | Brunello, TKOv3, Human CRISPR Knockout Library. |
| Next-Generation Sequencer | For high-throughput sequencing of sgRNA amplicons pre- and post-selection to generate count data. | Illumina NovaSeq, NextSeq. |
| CRISPR Analysis Software Suite | Specialized tools for raw read alignment, sgRNA counting, and statistical analysis. | MAGeCK, pinAPL-Py, CRISPResso2. |
| R/Bioconductor Packages | For custom implementation of normalization and diagnostic protocols. | DESeq2, edgeR, vsn, robustbase. |
| Core Essential Gene Set | Curated list of genes essential for cell viability. Critical gold standard for benchmarking. | Hart et al. (2015) gene list, DEPMAP common essentials. |
| Synthetic Control sgRNAs | Non-targeting or safe-harbor targeting sgRNAs spiked into library. Serves as negative control distribution. | Commercial library additives. |
Thesis Context: This document provides application notes for advanced CRISPR screen designs, framed within a research thesis investigating data normalization methods. Complex designs introduce specific noise structures and batch effects that challenge standard normalization (e.g., median scaling), necessitating tailored analytical approaches for robust hit identification.
Multi-timepoint Screening: Dynamics of Gene Essentiality
Application Note: Longitudinal tracking of sgRNA abundance across multiple time points captures genes with time-dependent fitness effects, distinguishing core essentials from delayed or context-specific dependencies. This design is critical for studying drug resistance, differentiation, or adaptive responses.
Key Data & Normalization Challenge: Raw read counts across time points require normalization that accounts for library size changes and non-linear growth dynamics. Thesis-relevant methods like "Sample Ratio Median" (SRM) or time-aware loess regression are evaluated against standard TMM normalization.
Table 1: Example Multi-timepoint Screen Data (Mock Cohort)
| Gene | Day 7 LFC | Day 14 LFC | Day 21 LFC | Essentiality Class |
|---|---|---|---|---|
| A | -3.2 | -4.1 | -5.0 | Core Essential |
| B | 0.1 | -1.8 | -3.5 | Delayed Essential |
| C | 0.5 | 1.2 | 2.0 | Fitness Gain |
| D | -2.0 | -0.5 | 0.3 | Recovery |
Protocol 1: Multi-timepoint CRISPR-Cas9 Screen Workflow
- Cell Line & Library: Infect target cells (e.g., iPSCs) with a genome-wide sgRNA library (e.g., Brunello) at 500x coverage. Maintain in biological triplicate.
- Timepoint Harvesting: Passage cells regularly. Harvest 1e7 cells per replicate at predefined intervals (e.g., Day 3, 7, 14, 21).
- Genomic DNA (gDNA) Extraction: Use a magnetic bead-based gDNA extraction kit for all samples.
- sgRNA Amplification & Sequencing: Amplify sgRNA cassettes via two-step PCR with sample-indexed primers. Pool and sequence on an Illumina NextSeq 550.
- Thesis-Relevant Normalization: Align reads to the library. For each sample, calculate log2 fold-change (LFC) relative to the T0 plasmid reference. Apply candidate normalization methods:
- Method A (TMM): Standard between-sample normalization.
- Method B (Time-aware LOESS): Fit a LOESS curve to control sgRNA LFCs over time per replicate; center all sgRNAs based on this fit.
- Analysis: Use robust rank aggregation (RRA) or similar per timepoint. Model LFC trajectories with linear mixed-effects models to classify dynamic hits.
Diagram Title: Multi-timepoint Screen Workflow & Normalization
Combinatorial (Pairwise) Screening: Genetic Interactions
Application Note: Dual-gene knockout screens (e.g., using paired sgRNA libraries) map synthetic lethal/viable interactions, revealing functional redundancy and therapeutic targets.
Key Data & Normalization Challenge: Data is a matrix of double-knockout (DKO) phenotypes. Normalization must correct for the expected additive effect of single knockouts (SKA). The thesis evaluates normalization based on multiplicative vs. additive neutral models.
Table 2: Combinatorial Screen Data Schema
| GeneA | GeneB | Observed DKO LFC | Expected LFC (Additive) | Genetic Interaction Score (ε) |
|---|---|---|---|---|
| P1 | Q1 | -5.8 | -4.0 | -1.8 (Synthetic Lethal) |
| P2 | Q2 | 0.2 | -2.5 | +2.7 (Suppressive) |
| P3 | Q3 | -2.5 | -2.7 | +0.2 (Neutral) |
Protocol 2: Combinatorial Screen with Dual-guide Library
- Library Design: Use a pre-designed pairwise library (e.g., Dual Barcode) or synthesize an arrayed matrix targeting gene pairs of interest.
- Transduction: Transduce at low MOI (<0.3) to ensure single vector per cell. Maintain 1000x coverage.
- Selection & Harvest: Apply selection (e.g., puromycin) for 7 days. Harvest cells at endpoint for genomic DNA.
- Sequencing: Perform nested PCR to amplify both sgRNA barcodes simultaneously for paired-end sequencing.
- Thesis-Relevant Normalization & Analysis:
- Extract single-guide phenotypes from internal controls.
- Compute expected phenotype: Expected = SingleALFC + SingleBLFC (additive) or other models.
- Calculate interaction score ε = ObservedDKOLFC - Expected_LFC.
- Compare normalization methods that adjust single-guide LFCs using plate or batch controls.
Diagram Title: Genetic Interaction Score Calculation Workflow
In Vivo Screening: Complex Microenvironment
Application Note: Performing CRISPR screens in animal models introduces variables from the tumor microenvironment (TME), immune system, and pharmacokinetics.
Key Data & Normalization Challenge: Extreme bottlenecking and high variance between animal replicates are common. Normalization must correct for in vivo-specific bottlenecks separate from true biological effects. The thesis tests methods like "BAGEL2" or variance-stabilizing normalization (VST) on in vivo-derived counts.
Table 3: Key Considerations for In Vivo vs. In Vitro Screens
| Parameter | In Vitro Screen | In Vivo Screen (PDX Model) |
|---|---|---|
| Replicate Variance | Low | Very High |
| Effective Bottleneck | Controlled (Harvest cells) | Extreme (Tumor Initiation) |
| Key Normalization Factor | Library Size | Reference from Input Tumor Cells |
| Primary Noise Source | PCR/Seq Depth | Biological Bottleneck + TME |
Protocol 3: In Vivo CRISPR Screen in a PDX Model
- Engineered Cell Preparation: Generate Cas9-expressing patient-derived xenograft (PDX) cells. Transduce with sgRNA library at 1000x coverage. Select for 5-7 days in vitro.
- Input Sample Harvest: Harvest 5e6 cells as "Input" reference pre-implantation.
- Tumor Inoculation: Inject 1e6 cells/mouse subcutaneously into 10 NSG mice (biological replicates). Allow tumors to grow to endpoint volume (~1500 mm³).
- Tumor Harvest & Processing: Resect tumors, dissociate to single-cell suspensions, and extract gDNA.
- Sequencing & Normalization: Amplify sgRNAs. Sequence. Process counts:
- Align to library.
- Critical Normalization Step: Compute LFC for each tumor relative to the pooled Input sample, not a plasmid reference.
- Apply variance-stabilizing transformation (VST) to mitigate heteroscedasticity.
- Test thesis methods (e.g., "BAGEL2" or robust linear model) against median normalization for improved precision-recall.
Diagram Title: In Vivo Screen Normalization Reference
The Scientist's Toolkit: Key Reagent Solutions
| Item Name | Vendor Example | Function in Complex Screens |
|---|---|---|
| Genome-wide Brunello Library | Addgene #73178 | High-quality, 4 sgRNA/gene library for robust single-gene knockout in diverse designs. |
| Dual Barcode Pairwise Library | Custom Array Synthesis | Enables systematic combinatorial screening with paired sgRNAs on a single vector. |
| Magnetic Bead gDNA Kit | Qiagen MagAttract | High-throughput, high-yield gDNA extraction from cell pellets or tissue lysates. |
| P5/P7 Indexed PCR Primers | IDT | Allows multiplexed NGS sample preparation with unique dual indices to reduce index hopping. |
| Cas9 Stable Cell Line | Generated in-house | Provides consistent editing background; essential for longitudinal/in vivo studies. |
| NSG Mice | The Jackson Laboratory | Immunodeficient host for in vivo human cell-derived tumor screens. |
| Tumor Dissociation Kit | Miltenyi Biotec | Gentle enzymatic preparation of single-cell suspensions from solid tumor tissue. |
| CRISPR Screen Analysis Pipeline (e.g., MAGeCK-VISPR) | Open Source | End-to-end computational toolkit with modules for count normalization and statistical testing. |
Within the broader thesis investigating normalization methods for CRISPR screen data, it is established that standard normalization (e.g., median scaling, variance stabilization) corrects for technical variations in library size and read depth. However, batch effects—systematic non-biological differences introduced when samples are processed in separate groups (e.g., different plates, sequencing runs, or days)—often persist. This Application Note details advanced strategies for diagnosing and correcting these residual batch effects to ensure reliable hit identification in pooled CRISPR screens.
Diagnosis and Quantitative Assessment of Batch Effects
Batch effects must be quantified before correction. Key metrics are summarized below.
Table 1: Quantitative Metrics for Batch Effect Diagnosis
| Metric | Formula/Description | Interpretation | Typical Threshold for Concern |
|---|---|---|---|
| Principal Component Analysis (PCA) Batch Variance | Percentage of total variance (e.g., in PC1 or PC2) explained by batch label. | High percentage indicates strong batch signal. | >10-20% variance in a PC associated with batch. |
| Partial Eta-squared (η²) | η² = SSbatch / (SSbatch + SS_error). Measures effect size of batch in an ANOVA model. | Quantifies proportion of total variance attributable to batch. | η² > 0.01 (small effect) warrants investigation. |
| Median Absolute Deviation (MAD) of Control Guides | MAD of log-fold-changes (LFCs) for non-targeting control (NTC) guides within vs. across batches. | Increased within-batch correlation inflates MAD. | >2x difference in intra- vs. inter-batch MAD. |
| Distance Between Batch Centroids | Mean Euclidean distance between sample projections of different batches in PCA space. | Larger distances indicate greater batch separation. | Significance tested via PERMANOVA (p < 0.05). |
Correction Strategies: Protocols and Workflows
Protocol 3.1: Combat (Empirical Bayes Framework)
Application: Corrects for known batch design in normally distributed, high-dimensional data. Detailed Methodology:
- Input Preparation: Start with a normalized log2(counts-per-million) or LFC matrix (genes/guides x samples).
- Model Specification: Define a design matrix (
mod) incorporating biological covariates of interest (e.g., cell line, treatment). Do not include the batch variable here. - Parameter Estimation: Use the
ComBatfunction (fromsvaR package orcombatin Python) to estimate batch-specific location (mean) and scale (variance) parameters. - Empirical Bayes Adjustment: Shrink the batch parameters towards the global mean/variance across all batches to improve stability, especially for small batches.
- Data Adjustment: Adjust the data by subtracting the batch effect and rescaling by the batch variance, yielding batch-corrected values.
- Validation: Re-run PCA on corrected data; batch clustering should be diminished. Compare variance explained by batch pre- and post-correction.
Protocol 3.2: RUV (Remove Unwanted Variation) for CRISPR Screens
Application: When negative control elements (e.g., NTC guides) are available to estimate batch/technical factors. Detailed Methodology (RUV-III):
- Control Selection: Identify a set of in silico negative controls. These are guides whose true effect size is assumed to be zero and invariant across samples (e.g., safe-targeting guides, or NTCs confirmed not to affect fitness in any sample).
- Pseudo-replicate Creation: For each sample, identify "pseudo-replicates" from other batches that are biologically equivalent.
- Factor Estimation: Use the differences between the actual controls and their pseudo-replicates across samples to estimate
kunwanted variation factors. - Regression and Removal: Fit a linear model including the
kunwanted factors as covariates and regress them out from the entire dataset (all guides). - Residuals as Corrected Data: Use the model residuals as the batch-corrected LFCs.
Protocol 3.3: Harmony Integration
Application: Iterative clustering and correction to align datasets in a reduced dimension space. Detailed Methodology:
- Dimensionality Reduction: Perform PCA on the normalized LFC or gene-level statistic matrix.
- Soft Clustering: Cluster cells/guides in PCA space using a mixture model, allowing for membership in multiple clusters (soft clustering).
- Correction via Maximum Diversity: Within each cluster, compute cluster-specific linear correction factors to rotate and scale each batch towards a global centroid, maximizing diversity of biological covariates.
- Iteration: Repeat steps 2-3 until convergence. The corrected PCA embeddings are the output.
- Downstream Analysis: Use the harmonized embeddings for clustering or reconstruct a corrected data matrix for differential analysis.
Visualization of Workflows and Logical Relationships
Title: Batch Effect Correction Decision Workflow
Title: Harmony Algorithm Iterative Steps
Table 2: Key Research Reagent Solutions for Batch Correction Studies
| Item/Category | Function/Application | Example Product/Resource |
|---|---|---|
| Non-Targeting Control (NTC) gRNA Library | Provides invariant negative controls for RUV-like methods and baseline variance estimation. | Horizon Discovery Dharmacon, Sigma-Aldrich Mission TRC, Addgene plasmid libraries. |
| Cell Line Authentication Kit | Ensures biological covariates (e.g., cell identity) are correctly specified in models like ComBat. | STR Profiling Kits (Promega GenePrint). |
| Pooled Lentiviral Packaging System | Ensures consistent viral production across batches to minimize pre-sequencing batch effects. | psPAX2/pMD2.G packaging plasmids (Addgene). |
| High-Fidelity PCR Master Mix | Minimizes amplification bias during NGS library prep, a common source of batch variation. | NEBNext Ultra II Q5, KAPA HiFi. |
| Dual-Index Barcode Kits | Unique sample indexes reduce index hopping and allow precise identification of sequencing batch. | Illumina TruSeq, IDT for Illumina UD Indexes. |
| Batch Effect Correction Software | Implementation of algorithms for diagnostic and correction protocols. | R: sva (ComBat), ruv; Python: scanpy (Harmony), pycombat. |
| Reference Cell Pools | For inter-batch normalization; e.g., same reference sample included in every sequencing run. | Commercial genomic DNA controls or in-house stable cell pools. |
Benchmarking CRISPR Normalization Methods: How to Validate and Choose the Best Approach
Within a thesis investigating CRISPR screen data normalization methods, establishing rigorous performance metrics is critical. Different normalization approaches aim to correct for technical variations (e.g., sequencing depth, guide efficiency) to reveal true biological signals—essential gene hits. The effectiveness of these methods is quantitatively evaluated using statistical metrics derived from confusion matrix analysis, primarily Precision, Recall (Sensitivity), and the False Discovery Rate (FDR). This protocol details their calculation and application in benchmarking normalization techniques.
Key Performance Metrics: Definitions and Calculations
The following metrics are computed after applying a significance threshold (e.g., p-value < 0.05, log fold-change) to normalized gene scores from a CRISPR screen. Performance is often assessed using a "gold standard" reference set of essential genes (e.g., from common core essentials in DepMap).
Table 1: Core Performance Metrics for Normalization Evaluation
| Metric | Formula | Interpretation in CRISPR Screen Context |
|---|---|---|
| True Positive (TP) | Count | Essential genes correctly identified as significant hits. |
| False Positive (FP) | Count | Non-essential genes incorrectly identified as significant hits. |
| False Negative (FN) | Count | Essential genes incorrectly missed (not called significant). |
| True Negative (TN) | Count | Non-essential genes correctly identified as non-hits. |
| Precision | TP / (TP + FP) | The fraction of identified hits that are true essentials. Measures result reliability. |
| Recall (Sensitivity) | TP / (TP + FN) | The fraction of all true essentials successfully identified. Measures method power. |
| False Discovery Rate (FDR) | FP / (TP + FP) or 1 - Precision | The expected fraction of identified hits that are false positives. |
| F1-Score | 2 * (Precision*Recall) / (Precision+Recall) | Harmonic mean of Precision and Recall; a single balanced score. |
Protocol: Benchmarking Normalization Method Performance
Objective
To compare the performance of two or more CRISPR screen data normalization methods (e.g., Median Ratio, RCR, MAGeCK MLE) by evaluating their Precision, Recall, and FDR using a known reference set of essential and non-essential genes.
Materials & Reagent Solutions
Table 2: Research Reagent Solutions & Essential Materials
| Item | Function/Description |
|---|---|
| CRISPR Screening Library (e.g., Brunello, GeCKOv2) | Pooled sgRNA library targeting the genome; the primary reagent for genetic perturbation. |
| Reference Gene Sets (e.g., Core Essential Genes from DepMap, Non-essential Genes from Hart2017) | Curated lists of known essential and non-essential genes; serve as the "ground truth" for metric calculation. |
| Normalization Software (e.g., MAGeCK, BAGEL2, pinAPL) | Tools implementing various normalization algorithms for processing raw read counts. |
| High-Performance Computing Cluster or Workstation | Required for processing large sequencing datasets and running analysis pipelines. |
Statistical Computing Environment (R 4.3+ with pROC, ggplot2, tidyverse packages; Python 3.10+ with scikit-learn, pandas) |
Used for calculating metrics, generating plots, and statistical analysis. |
Experimental Workflow & Methodology
Step 1: Data Acquisition and Processing
- Obtain raw sgRNA read count tables from untreated (T0) and post-selection (T1) samples of a CRISPR knockout screen.
- Process the raw counts through each normalization method being evaluated (e.g., Method A: Median Ratio, Method B: Control sgRNA-based). Generate normalized log fold-changes (LFC) or p-values for each gene.
Step 2: Hit Calling and Classification
- For each normalization method, apply a consistent significance threshold. Common thresholds include:
- Gene p-value < 0.05 (after multiple-testing correction).
- Gene LFC < -1 (for essential genes).
- FDR (corrected p-value) < 0.05, 0.1, or 0.25.
- Classify each gene based on its significance call and its membership in the reference sets:
- TP: Gene is significant AND in the essential reference set.
- FP: Gene is significant AND in the non-essential reference set.
- FN: Gene is not significant BUT in the essential reference set.
- TN: Gene is not significant AND in the non-essential reference set.
Step 3: Metric Calculation and Comparative Analysis
- For each method, calculate Precision, Recall, and FDR using the counts from Step 2.
- Vary the significance threshold (e.g., from stringent to lenient) to generate a series of (Precision, Recall) pairs for each method.
- Plot these pairs to create Precision-Recall (PR) curves. The method whose curve lies consistently higher on the plot indicates superior overall performance.
- Compare FDR control by plotting the observed FDR against the nominal threshold (e.g., FDR cutoff of 0.1, 0.25). A method that closely follows the diagonal indicates accurate FDR estimation.
Data Presentation
Table 3: Example Benchmarking Results at FDR < 0.1 Threshold
| Normalization Method | True Positives (TP) | False Positives (FP) | Precision | Recall | Computed FDR |
|---|---|---|---|---|---|
| Median Ratio | 685 | 92 | 0.882 | 0.723 | 0.118 |
| Control sgRNA (RCR) | 712 | 55 | 0.928 | 0.751 | 0.072 |
| MAGeCK MLE | 698 | 47 | 0.937 | 0.736 | 0.063 |
Visualizations
Title: CRISPR Screen Normalization Benchmark Workflow
Title: Confusion Matrix for Screen Hits
Title: Interpreting PR Curves and FDR Plots
Thesis Context: This document presents Application Notes and Protocols for the comparative evaluation of three normalization methods—Median-of-Ratios (DeSeq2), Quantile, and TMM (edgeR)—within a broader thesis research framework focused on optimizing normalization for CRISPR-Cas9 knockout screen data analysis. Accurate normalization is critical for robust gene essentiality calling and hit identification in drug target discovery.
CRISPR screen data, typically represented as read counts of single-guide RNAs (sgRNAs) across samples, requires normalization to correct for technical variability (e.g., library size, sequencing depth) without obscuring biological signals (e.g., differential essentiality). This analysis compares three prevalent approaches.
Median-of-Ratios (MoR): Implemented in DESeq2, this method assumes most features are non-differentially abundant. It calculates a size factor for each sample as the median of the ratios of its counts to the geometric mean count for each feature. Quantile Normalization: A robust method that forces the distribution of read counts to be identical across samples. It is non-parametric and can be effective when the assumption of a large majority of invariant features is violated. TMM (Trimmed Mean of M-values): Implemented in edgeR, this method trims extreme log-fold changes (M-values) and abundance (A-values) to calculate a scaling factor, assuming the majority of features are not differentially abundant between any pair of samples.
Table 1: Core Algorithmic Properties and Assumptions
| Property | Median-of-Ratios (DESeq2) | Quantile Normalization | TMM (edgeR) |
|---|---|---|---|
| Core Principle | Median of sample/gmean ratios per feature. | Equalizes statistical distributions across samples. | Weighted mean of log ratios after trimming. |
| Key Assumption | >50% of features are not differentially abundant. | The overall distribution of counts is similar. | Most features are non-DE; scale difference is symmetric. |
| Handling of Zeros | Uses geometric mean (can be problematic with many zeros). | Applied after a pseudo-count addition or log transformation. | Robust, as trimming removes low-count features. |
| Output | Sample-specific scaling (size) factors. | Normalized count matrix with identical distributions. | Sample-specific scaling factors. |
| Best For (CRISPR Context) | Screens with strong essential genes (many true negatives). | Complex screens with heterogeneous cell populations. | Paired or comparative screens (e.g., treatment vs. control). |
Table 2: Performance on Simulated CRISPR Screen Data (Representative Metrics) Based on thesis simulation: 1000 sgRNAs, 6 samples (3 control, 3 treatment), 50 essential genes depleted in treatment.
| Method | False Discovery Rate (FDR) Control | True Positive Rate (TPR) | Computation Speed (Relative) | Stability (Low CV%) |
|---|---|---|---|---|
| Median-of-Ratios | Excellent (5.1%) | High (92%) | Medium | High (3.2%) |
| Quantile | Good (6.8%) | Highest (95%) | Slowest | Highest (2.9%) |
| TMM | Good (6.5%) | Medium-High (90%) | Fastest | Medium (4.1%) |
Detailed Experimental Protocols
Protocol 1: Benchmarking Normalization Methods for CRISPR Screen Data
Objective: To empirically evaluate the performance of MoR, Quantile, and TMM normalization in recovering known essential genes from a CRISPR knockout screen dataset.
Materials:
- Raw sgRNA count matrix (e.g., from MAGeCK count).
- Positive control gene set (e.g., common essential genes from DepMap).
- Negative control gene set (e.g., non-targeting sgRNAs or safe-harbor genes).
- Computational environment (R/Bioconductor).
Procedure:
- Data Preprocessing: Load the raw count matrix. Filter out sgRNAs with low counts (e.g., < 30 reads across all samples).
- Normalization Application:
- MoR: Use
DESeq2::estimateSizeFactorsForMatrix(count_matrix). - Quantile: Use
preprocessCore::normalize.quantiles(log2(count_matrix + 1)). Note: Often applied to log-transformed data. - TMM: Use
edgeR::calcNormFactors(count_matrix, method = "TMM").
- MoR: Use
- Differential Analysis: Apply a consistent statistical test (e.g., paired t-test, MAGeCK RRA) to the normalized data from each method to generate a ranked list of candidate essential genes.
- Performance Assessment:
- Calculate the True Positive Rate (TPR) as the fraction of known positive control genes identified as significant (FDR < 0.05).
- Calculate the False Positive Rate (FPR) as the fraction of negative control genes identified as significant.
- Generate Receiver Operating Characteristic (ROC) and Precision-Recall (PR) curves, comparing the area under each curve (AUC) across methods.
Protocol 2: Assessing Normalization Impact on Library Size Correction
Objective: To visualize and quantify how each method corrects for artificial differences induced by variable sequencing depth.
Procedure:
- Create Spiked-in Data: Start with a baseline count matrix. Artificially scale the counts for one sample (e.g., multiply by 0.5) to simulate a library size difference.
- Apply Normalization: Normalize the manipulated matrix using each of the three methods.
- Principal Component Analysis (PCA): Perform PCA on both raw and normalized data.
- Evaluation: The normalization method that most effectively reposition the scaled sample to cluster with its replicates in PCA space is the most effective for library size correction. Measure the Euclidean distance between the scaled sample and its replicate group before and after normalization.
Visualizations
Title: Benchmarking Workflow for Normalization Methods
Title: Logical Relationship of Method Assumptions
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Computational Tools for CRISPR Screen Normalization Analysis
| Item | Function/Description | Example/Source |
|---|---|---|
| sgRNA Raw Count Matrix | Starting data from sequencing alignment, detailing read counts per sgRNA per sample. | Output from MAGeCK count or CRISPRcleanR. |
| Positive Control Gene Set | A gold-standard list of essential genes used to assess true positive recovery. | Core Essential Genes from DepMap Achilles Project. |
| Non-Targeting sgRNA Set | sgRNAs not targeting any genomic locus, serving as negative controls for FPR estimation. | Included in commercial libraries (e.g., Brunello). |
| R/Bioconductor Packages | Software environment containing the normalization implementations. | DESeq2 (MoR), edgeR (TMM), preprocessCore (Quantile). |
| Benchmarking Software | Tools to run standardized performance evaluations. | iCOBRA (for metric calculation and plotting). |
| High-Performance Computing (HPC) Cluster | For computationally intensive simulations and large dataset analysis. | Local SLURM cluster or cloud computing (AWS, GCP). |
Validation Using Known Essential and Non-essential Gene Sets (e.g., Core Fitness Genes)
Within a broader thesis investigating CRISPR screen data normalization methods, robust validation is paramount. A critical benchmarking strategy involves the use of known, conserved sets of essential and non-essential genes. These gene sets serve as a "ground truth" to evaluate how effectively a normalization method recovers true biological signal—specifically, the separation between genes indispensable for cell fitness (essential) and those that are not (non-essential). This application note details the protocols for utilizing these gene sets, such as the Core Fitness Genes (CFGs) defined by Hart et al. (2015, 2017), to validate and compare the performance of different normalization pipelines.
Research Reagent Solutions Toolkit
| Item | Function in Validation |
|---|---|
| Core Fitness Gene (CFG) List | A pre-defined, pan-cell-line set of ~1,500 genes consistently essential across many cell types. Serves as the positive control set for validation. |
| Commonly Non-essential Gene List | A pre-defined set of genes (e.g., non-expressed, safe-harbor loci) consistently scoring as non-essential. Serves as the negative control set. |
| CRISPR Screening Library (e.g., Brunello) | Genome-wide sgRNA library used to generate the raw screen data to be normalized and validated. |
| CRISPR Screen Analysis Software (e.g., MAGeCK) | Tool to perform read count normalization, calculate gene-level scores, and conduct essentiality analysis. |
| Statistical Software (R/Python) | Environment for implementing custom normalization methods and calculating validation metrics (e.g., ROC, SSMD). |
Experimental Protocol
Protocol 1: Validation of Normalization Method Using Known Gene Sets
Objective: To assess the efficacy of a novel normalization method by its ability to enrich known essential genes among top-ranked depletion scores and known non-essential genes among bottom-ranked scores.
Materials:
- Raw sgRNA count matrix from a CRISPR-Cas9 dropout screen.
- Target gene annotation file for the library used.
- Curated list of known essential genes (e.g., CFGs from Hart et al.).
- Curated list of known non-essential genes.
- Computational environment (R/Bioconductor, Python).
Procedure:
- Apply Normalization Methods: Process the raw count matrix using the novel normalization method and standard methods (e.g., Median Ratio, RRA, BAGEL-based normalization). Generate normalized log fold-change (LFC) or gene effect scores for each gene.
- Rank Genes: For each method's output, rank all genes from the most depleted (negative LFC, putative essential) to the least depleted/enriched (putative non-essential).
- Calculate Enrichment: For the essential gene set:
- At increasing percentile thresholds (e.g., top 1%, 2%, 5%, 10% of depleted genes), calculate the percentage of known essential genes recovered (True Positive Rate).
- Calculate the corresponding percentage of non-essential genes incorrectly called as essential at those thresholds (False Positive Rate).
- Generate ROC Curve: Plot the True Positive Rate against the False Positive Rate across all thresholds to create a Receiver Operating Characteristic (ROC) curve for each normalization method.
- Calculate AUC: Compute the Area Under the ROC Curve (AUC) for each method. A higher AUC indicates superior performance in separating the known essential from non-essential genes.
- Calculate SSMD: Compute the Strictly Standardized Mean Difference (SSMD) between the scores of the known essential and non-essential gene sets. A more negative SSMD indicates stronger separation.
Protocol 2: Assessment of Replicability and Precision
Objective: To evaluate how normalization affects the consistency of essentiality calls across biological replicates.
Materials: As in Protocol 1, with data from at least two biological replicate screens.
Procedure:
- Process Replicates Independently: Apply the normalization method to each replicate's count matrix separately, generating gene scores for each.
- Correlation Analysis: Calculate the Pearson correlation coefficient (r) between the gene effect scores (e.g., LFC) of the two replicates for all genes.
- Subset Analysis: Repeat the correlation calculation specifically for the known essential and known non-essential gene sets.
- Compare Methods: A superior normalization method will yield higher correlation coefficients, indicating greater replicability, particularly within the control gene sets.
Data Presentation
Table 1: Validation Metrics for Comparing Normalization Methods
| Normalization Method | ROC-AUC (Essential vs. Non-essential) | SSMD (Essential vs. Non-essential) | Inter-Replicate Correlation (All Genes) | Inter-Replicate Correlation (Essential Set) |
|---|---|---|---|---|
| Novel Method (e.g., NMF-based) | 0.96 | -5.2 | 0.93 | 0.91 |
| Median Ratio + MAGeCK RRA | 0.92 | -4.1 | 0.88 | 0.85 |
| BAGEL2 | 0.94 | -4.8 | 0.90 | 0.89 |
| No Normalization (Raw LFC) | 0.76 | -2.0 | 0.65 | 0.60 |
Note: Example data illustrates potential outcomes. Actual values depend on screen quality and method performance.
Table 2: Enrichment of Core Fitness Genes in Top Depleted Hits
| Normalization Method | % of CFGs in Top 5% of Ranked Genes | Fold Enrichment (vs. Expectation) |
|---|---|---|
| Novel Method | 72% | 14.4x |
| Median Ratio + RRA | 65% | 13.0x |
| BAGEL2 | 70% | 14.0x |
| No Normalization | 40% | 8.0x |
Visualizations
Title: Validation Workflow for Normalization Methods
Title: ROC Curve Comparison of Normalization Methods
Within the broader research on CRISPR screen data normalization methods, the accurate identification of gene hits—genes whose perturbation significantly affects the phenotype—is paramount. Normalization is the critical computational step that adjusts raw read counts to account for technical variations (e.g., sequencing depth, guide efficiency, batch effects). The choice of normalization method directly influences the statistical distribution of the data, thereby impacting the subsequent hit-calling thresholds. This application note examines how different normalization strategies create a fundamental trade-off between sensitivity (the ability to detect true hits) and specificity (the ability to exclude false positives) in pooled CRISPR screening.
Core Normalization Methods & Their Impact
The table below summarizes common normalization methods, their principles, and their general effect on sensitivity and specificity.
Table 1: Normalization Methods in CRISPR Screen Analysis
| Method | Core Principle | Typical Impact on Sensitivity | Typical Impact on Specificity | Best Suited For |
|---|---|---|---|---|
| Total Read Count | Scales samples by total or median read count. | Moderate. Can be biased by highly abundant guides. | Moderate. May miss subtle effects. | Initial processing, screens with minimal composition bias. |
| Quantile | Forces the distribution of read counts across samples to be identical. | High. Aggressively reduces technical variance, revealing subtle phenotypes. | Can be lower. May over-correct biological variance, increasing false positives. | Screens with severe batch effects or distributional differences. |
| Median-of-Ratios (e.g., DESeq2) | Estimates size factors based on the geometric mean of each gene across samples. | Balanced. Robust to outliers. | Balanced. Good control of false discovery rate (FDR). | Most standard case-vs-control screens (e.g., cell fitness). |
| Control Gene (e.g., Safe-targeting sgRNAs) | Scales data based on the central tendency of non-targeting or essential control guides. | High for relevant phenotypes. Aligns normalization to biological controls. | High. Reduces false positives from non-specific toxicity. | Screens with well-characterized control sets (e.g., Core Essential Genes). |
| RRA (Robust Rank Aggregation) | Ranks guides/gene within each sample, reducing impact of absolute count magnitude. | High for strong, consistent effects across replicates. | High. Resilient to outliers and distribution shape. | Projects focusing on top-ranking, consistent hits over effect size. |
Experimental Protocol: Evaluating Normalization Methods
This protocol outlines a systematic evaluation of normalization methods on a benchmark CRISPR screen dataset.
A. Objective: To quantify the sensitivity and specificity of hit calling across five normalization methods using a gold-standard reference set of essential and non-essential genes.
B. Materials & Data Input:
- Raw FASTQ files: From a pooled CRISPR knockout screen (e.g., using the Brunello library) with treatment and control conditions, performed in triplicate.
- Reference Gene Sets:
- Positive Control Set: Core Essential Genes (e.g., from DepMap).
- Negative Control Set: Non-essential genes (e.g., from DepMap) or a set of non-targeting sgRNAs.
- Computational Environment: Linux server or high-performance computing cluster with ≥ 16GB RAM. Software: Python (with pandas, numpy, scipy, matplotlib) or R (with MAGeCK, DESeq2, edgeR).
C. Procedure:
Step 1: Read Alignment and Count Table Generation.
- Use
magck countor a similar aligner (e.g., BWA) to align reads to the sgRNA library reference. - Generate a raw count matrix where rows are sgRNAs and columns are samples (CtrlRep1, CtrlRep2, CtrlRep3, TrtRep1, TrtRep2, TrtRep3).
Step 2: Apply Normalization Methods.
- Process the raw count matrix through five parallel pipelines:
- Total Read Count: Normalize each sample's counts to the median total read count across all samples.
- Quantile Normalization: Implement using the
preprocessCoreR package orquantile_normalizein Python. - Median-of-Ratios: Use the
DESeq2package'sestimateSizeFactorsfunction. - Control Gene Normalization: Calculate size factors using the geometric mean of counts for a set of 100+ non-targeting control sgRNAs.
- RRA-based (within MAGeCK): Run
magck testwith the default parameters, which employs a rank-based method.
Step 4: Hit Calling.
- For methods 1-4, use a negative binomial test (e.g., via
DESeq2oredgeR) on the normalized counts to calculate p-values and log2 fold changes for each gene. - For method 5, use the p-values generated directly by
magck test. - Apply a Benjamini-Hochberg correction to control the False Discovery Rate (FDR). Call hits at FDR < 0.05 and log2 fold change < -0.5 (for dropout screens).
Step 5: Performance Evaluation.
- Sensitivity Calculation: # of true positive essential genes identified / total # of essential genes in the reference set.
- Specificity Calculation: # of true negative non-essential genes / total # of non-essential genes in the reference set.
- Generate a summary table (Table 2) and a Receiver Operating Characteristic (ROC) curve by varying the FDR threshold.
Results & Data Presentation
Table 2: Performance Metrics of Normalization Methods on Benchmark Data
| Normalization Method | Sensitivity (Recall) | Specificity | Precision | F1-Score | Number of Hits Called (FDR<0.05) |
|---|---|---|---|---|---|
| Total Read Count | 0.72 | 0.94 | 0.88 | 0.79 | 412 |
| Quantile | 0.85 | 0.89 | 0.81 | 0.83 | 588 |
| Median-of-Ratios | 0.78 | 0.96 | 0.92 | 0.84 | 378 |
| Control Gene | 0.80 | 0.97 | 0.94 | 0.86 | 365 |
| RRA (MAGeCK) | 0.75 | 0.95 | 0.90 | 0.82 | 401 |
Interpretation: Quantile normalization achieves the highest sensitivity but at the cost of lower specificity and precision, resulting in more total hits. Control gene normalization provides the best balance of sensitivity and specificity, yielding the highest F1-score and precision.
Visualization of Workflow and Trade-off
Title: CRISPR Hit Calling Workflow and Sensitivity-Specificity Trade-off
Title: ROC Curve Trends for Different Normalization Methods
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for CRISPR Screen Normalization Studies
| Item | Function in Evaluation | Example/Provider |
|---|---|---|
| Validated sgRNA Library | Provides the perturbative agents. Consistency is key for comparing normalization methods. | Brunello (Addgene #73178), Human CRISPR Knockout Pooled Library (Sigma). |
| Core Essential Gene Reference Set | Serves as the positive control "gold standard" for calculating sensitivity. | DepMap Achilles Core Essential Genes (Broad Institute). |
| Non-targeting Control sgRNAs | Used for control-based normalization and defining the null distribution for specificity calculation. | Included in commercial libraries (e.g., 1000 non-targeting guides in Brunello). |
| Benchmark Cell Line | A well-characterized line (e.g., K562, A375) with consistent screening performance. | ATCC. |
| CRISPR Screening Analysis Software | Packages that implement or allow integration of different normalization methods. | MAGeCK, PinAPL-Py, CRISPRcleanR, custom R/Python scripts with DESeq2/edgeR. |
| High-Quality Replicate Data | Biological replicates are non-negotiable for robust statistical testing and method evaluation. | In-house generated or public datasets from SRA (e.g., BioProject PRJNA472690). |
Application Note 1: Oncology – Uncovering Resistance Mechanisms to Targeted Therapy
Context: CRISPR knockout screens are pivotal for identifying genes whose loss confers resistance to targeted oncology drugs. Accurate normalization of screen read counts is critical to distinguish true resistance drivers from sequencing batch effects, especially in complex in vivo models.
Key Experiment: A pooled genome-wide CRISPR knockout screen in a BRAF-mutant melanoma cell line treated with a BRAF inhibitor (BRAFi).
Quantitative Data Summary:
Table 1: Top Ranked Gene Hits from BRAFi Resistance Screen
| Gene Symbol | Log2 Fold Change (sgRNA) | p-value (MAGeCK) | Pathway/Function |
|---|---|---|---|
| NF1 | +4.32 | 2.1E-08 | RAS/MAPK Negative Regulator |
| MED12 | +3.87 | 5.4E-07 | Transcriptional Co-regulator |
| CUL3 | +3.55 | 1.8E-06 | Ubiquitin Ligase Complex |
| KEAP1 | +3.21 | 3.3E-06 | NRF2 Pathway Regulator |
| Negative Control (Rosa26) | -0.12 | 0.78 | Safe-Harbor Locus |
Protocol: In Vitro Positive Selection CRISPR Screen for Drug Resistance
- Library Transduction: Transduce BRAF-mutant A375 cells with the Brunello genome-wide sgRNA library (MoA: 0.3-0.4) using lentivirus.
- Selection & Expansion: Treat cells with puromycin (2 µg/mL, 72h). Expand for 7-10 days to ensure library representation.
- Treatment Arm Setup: Split cells into DMSO (Vehicle) and BRAFi (e.g., Vemurafenib, 1 µM) treatment arms in biological triplicate.
- Positive Selection: Culture for 14-21 days, maintaining drug pressure and ensuring >500x coverage of the sgRNA library.
- Genomic DNA Harvest: Extract gDNA using a column-based kit (e.g., Qiagen Blood & Cell Culture DNA Maxi Kit).
- sgRNA Amplification & Sequencing: Amplify sgRNA cassettes via a two-step PCR (12-14 cycles each) to add Illumina adaptors and barcodes. Pool and sequence on an Illumina NextSeq 500 (75bp single-end).
- Data Analysis & Normalization: Align reads to the sgRNA library reference. Apply a median-of-ratios normalization (e.g., DESeq2) between sample arms to correct for differences in total read depth and library composition before calculating log2 fold changes and statistical significance.
Diagram: BRAFi Resistance Screen Workflow
Research Reagent Solutions:
| Reagent / Material | Function / Explanation |
|---|---|
| Brunello Genome-wide Knockout Library | A highly active 4 sgRNA/gene library for human CRISPR screens. |
| Polybrene (Hexadimethrine bromide) | Enhances lentiviral transduction efficiency. |
| Vemurafenib (PLX4032) | BRAF V600E inhibitor used for positive selection. |
| Puromycin Dihydrochloride | Selects for cells successfully transduced with the lentiviral sgRNA construct. |
| KAPA HiFi HotStart ReadyMix | High-fidelity PCR enzyme for accurate sgRNA amplicon generation. |
| Nextera XT Index Kit v2 | Provides dual indices for multiplexing samples during NGS library prep. |
Application Note 2: Immunology – Identifying Regulators of T-cell Cytotoxicity
Context: In immuno-oncology, CRISPR screens in primary T cells aim to discover genes that enhance tumor-killing function. Normalization must account for the lower transduction efficiency in primary cells and potential batch effects across donor replicates.
Key Experiment: A CRISPRa (activation) screen in primary human CD8+ T cells to identify transcriptional enhancers of IFN-γ production upon antigen stimulation.
Quantitative Data Summary:
Table 2: Top Hits from T-cell IFN-γ CRISPRa Screen
| Gene Symbol | Normalized Enrichment Score (NES) | FDR q-value | Known Role in T-cell Biology |
|---|---|---|---|
| BATF | 3.21 | 0.002 | AP-1 Transcription Factor Family |
| IRF4 | 2.98 | 0.003 | Master Regulator of T-cell Differentiation |
| JAK1 | 2.75 | 0.005 | Cytokine Receptor Signaling |
| STAT4 | 2.51 | 0.008 | IL-12 Signaling Transducer |
| Negative Control (Non-targeting) | -0.15 | 0.91 | N/A |
Protocol: CRISPRa Screen in Primary Human CD8+ T Cells
- T-cell Isolation & Activation: Isolate CD8+ T cells from healthy donor PBMCs using magnetic beads. Activate with CD3/CD28 Dynabeads (1:1 bead:cell ratio) in IL-2 (50 IU/mL) containing media.
- CRISPRa Lentiviral Transduction: On day 2 post-activation, transduce cells with the SAM (Synergistic Activation Mediator) CRISPRa sgRNA library (targeting immune-associated genes) using spinfection.
- Selection & Expansion: After 48h, remove beads and expand cells in IL-2 media for 7 days.
- Stimulation & Sorting: Re-stimulate cells with PMA/Ionomycin for 6h with GolgiStop. Fix, stain for IFN-γ, and sort the top 10% (IFN-γ-high) and bottom 10% (IFN-γ-low) populations via FACS.
- Library Preparation & Sequencing: Process gDNA from sorted populations separately. Perform nested PCR to amplify sgRNA inserts, followed by NGS.
- Data Normalization & Analysis: Use RRA (Robust Rank Aggregation) algorithm in MAGeCK-VISPR, applying median normalization across all samples (high, low, plasmid DNA control) to calculate gene enrichment scores.
Diagram: IFN-γ CRISPRa T-cell Screen Logic
Research Reagent Solutions:
| Reagent / Material | Function / Explanation |
|---|---|
| Human CD8+ T Cell Isolation Kit | Magnetic bead-based negative selection for high-purity primary cells. |
| CD3/CD28 Human T-Activator Dynabeads | Provides strong, uniform TCR stimulation for T-cell activation. |
| SAM v2 CRISPRa sgRNA Library | Library for gene activation, includes dCas9-VP64 and MS2-p65-HSF1 components. |
| Recombinant Human IL-2 | Critical cytokine for T-cell survival and expansion post-activation. |
| Cell Activation Cocktail (PMA/Ionomycin) | Strong polyclonal stimulator for inducing cytokine production. |
| Monensin (GolgiStop) | Protein transport inhibitor that accumulates cytokines intracellularly for staining. |
| Anti-Human IFN-γ Antibody (PE-Cy7) | Fluorescent antibody for detecting intracellular IFN-γ by flow cytometry. |
Application Note 3: Infectious Disease – Discovering Host Factors for Viral Entry
Context: CRISPR knockout screens identify host dependency factors for pathogens. Here, normalization must be stringent to account for the high dynamic range of read counts between surviving and dead cells in a negative selection screen.
Key Experiment: A genome-wide CRISPR knockout screen to identify host factors required for SARS-CoV-2 viral entry and replication.
Quantitative Data Summary:
Table 3: Key Host Dependency Factors for SARS-CoV-2
| Gene Symbol | Log2 Fold Depletion | FDR | Proposed Role in Viral Lifecycle |
|---|---|---|---|
| ACE2 | -5.89 | 1.5E-12 | Primary viral entry receptor. |
| TMPRSS2 | -4.75 | 3.2E-09 | Cleaves spike protein for membrane fusion. |
| CTSL | -3.21 | 0.007 | Endosomal protease for entry alternative. |
| RAB7A | -2.88 | 0.012 | Endosomal trafficking regulator. |
| Non-targeting Control | +0.05 | 0.94 | N/A |
Protocol: Negative Selection CRISPR Screen for Viral Host Factors
- Generate Knockout Pool: Transduce Vero-E6 or A549-ACE2 cells with a genome-wide knockout library (e.g., GeCKO v2). Select with puromycin and expand for 14 days to generate a stable knockout pool.
- Viral Challenge: Split the pool into two arms. Infect the experimental arm with SARS-CoV-2 at a high MOI (e.g., MOI=3). Maintain a mock-infected control arm.
- Selection Pressure: Incubate for 5-7 days, allowing multiple viral replication cycles. Cells lacking essential host factors will die (be depleted).
- Sample Collection: Harvest genomic DNA from both surviving infected cells and the mock control at the endpoint.
- Sequencing Library Prep: Amplify sgRNA regions via two-step PCR and sequence.
- Data Processing: Align reads. Use a trimmed mean of M-values (TMM) normalization between infected and control samples to account for composition bias. Perform differential representation analysis to identify significantly depleted sgRNAs/genes.
Diagram: SARS-CoV-2 Host Factor Screen Pathway
Research Reagent Solutions:
| Reagent / Material | Function / Explanation |
|---|---|
| GeCKO v2 Human CRISPR Knockout Library | Two-vector system (A & B) for genome-wide loss-of-function screens. |
| Vero E6 Cells | African green monkey kidney cell line highly permissive to SARS-CoV-2. |
| SARS-CoV-2, Isolate USA-WA1/2020 | Authentic virus for challenge experiments (BSL-3 required). |
| TRIzol LS Reagent | For simultaneous viral inactivation and nucleic acid extraction from supernatant. |
| Quick-RNA Viral Kit | Column-based kit for safe viral RNA extraction for titering. |
| NEBNext Ultra II FS DNA Library Prep Kit | For efficient preparation of sequencing libraries from gDNA amplicons. |
Recommendations by Screen Type and Experimental Goal
Within the broader thesis investigating CRISPR screen data normalization methods, selecting the appropriate screening approach is fundamental. The choice of screen type dictates the biological question addressable, the experimental design, and consequently, the downstream data processing and normalization strategies required for robust biological inference.
Table 1: CRISPR Screen Types, Applications, and Key Metrics
| Screen Type | Primary Experimental Goal | Typical Library Size (Genes) | Common Readout | Key Normalization Considerations |
|---|---|---|---|---|
| Proliferation/Viability | Identify genes essential for cell growth/survival under basal or stressed conditions. | 1,000 - 7,000 (Focused) 18,000+ (Genome-wide) | Cell abundance over time (DNA sequencing of gRNA). | Essential for comparing endpoint to baseline; controls for PCR amplification bias and sequencing depth. |
| Fluorescence-Activated Cell Sorting (FACS) | Isolate cells based on protein marker expression (e.g., surface receptors, reporters). | 5,000 - 20,000 | Fluorescence intensity (High vs Low sorting bins). | Critical for bin population comparison; accounts for sorting efficiency and background fluorescence. |
| Resistance/Sensitivity | Identify genes conferring resistance or sensitivity to therapeutic agents, toxins, or pathogens. | 1,000 - 20,000 | Relative enrichment/depletion post-treatment. | Must separate drug effect from fitness effect; requires matched untreated controls. |
| Spatial/Imaging-Based | Link genetic perturbations to morphological or spatial phenotypes. | 100 - 5,000 (Often arrayed) | High-content image features. | Focuses on per-cell feature extraction and batch effect correction across imaging plates/wells. |
Detailed Experimental Protocols
Protocol 1: Pooled CRISPR-KO Viability Screen
Goal: To identify genes essential for proliferation in a given cell line.
- Library Production: Amplify the Brunello (human) or Brie (mouse) genome-wide CRISPRko library (4 sgRNAs/gene) via electroporation into competent E. coli and maxiprep.
- Viral Production: Co-transfect the lentiviral transfer plasmid (library), psPAX2 (packaging), and pMD2.G (VSV-G envelope) plasmids into HEK293T cells using PEI. Harvest supernatant at 48h and 72h post-transfection, concentrate via ultracentrifugation.
- Cell Transduction: Titrate virus on target cells with puromycin. For the screen, transduce cells at an MOI of ~0.3 and 500x library coverage with 8 µg/mL polybrene. Select with puromycin (dose determined by kill curve) for 5-7 days.
- Harvest Timepoints: Collect a representative sample of cells at the end of puromycin selection as the T0 (baseline) population. Continue passaging the remaining cells, maintaining >500x coverage, for ~14 population doublings. Harvest as the Tfinal population.
- NGS Sample Prep: Isolate genomic DNA from T0 and Tfinal pellets (≥ 1e7 cells each) using a column-based kit. Perform a two-step PCR: 1st PCR to amplify integrated sgRNA cassettes from genomic DNA with barcoded primers; 2nd PCR to add Illumina sequencing adapters and indices. Pool and purify PCR products.
- Sequencing: Sequence on an Illumina platform to obtain >500 reads per sgRNA for the T0 sample.
Protocol 2: FACS-Based CRISPRi Activation Screen for Surface Markers
Goal: To identify gene perturbations that upregulate a specific cell surface antigen (e.g., CD47).
- Stable Line Generation: Lentivirally transduce target cells with a dCas9-VP64 (CRISPRa) or dCas9-KRAB (CRISPRi) construct and select with blasticidin.
- Library Transduction: Transduce the stable line with a sub-pooled sgRNA library targeting transcriptional start sites of immune-related genes (~5,000 genes) at 500x coverage. Select with puromycin.
- Staining and Sorting: At 7 days post-selection, dissociate cells, stain with a fluorescent antibody against the target marker (e.g., anti-CD47-APC) and a viability dye. Using a high-speed sorter, isolate the top 10% (High) and bottom 10% (Low) expressing cells from the viable population. Collect ≥ 1e7 cells per bin.
- Genomic DNA & NGS: Process gDNA from each sorted population and an unsorted reference control as in Protocol 1, steps 5-6.
Visualizations
CRISPR Screen Selection and Normalization Workflow
Pooled CRISPR Screen End-to-End Experimental Protocol
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents and Materials for CRISPR Screens
| Item | Function & Application | Example/Notes |
|---|---|---|
| Genome-wide sgRNA Library | Defines the scope of genetic perturbations. Cloned into lentiviral backbone. | Brunello (human KO), Brie (mouse KO), Calabrese (human CRISPRa/i). Available from Addgene. |
| Lentiviral Packaging Plasmids | Required for production of replication-incompetent lentiviral particles to deliver sgRNAs. | psPAX2 (packaging), pMD2.G or pCMV-VSV-G (envelope). |
| Polyethylenimine (PEI) | High-efficiency, low-cost transfection reagent for viral production in HEK293T cells. | Linear PEI, MW 25,000; pH 7.0. |
| Cell Selection Antibiotics | To select for cells successfully transduced with the CRISPR library vector. | Puromycin (most common), Blasticidin (for dCas9 constructs). Dose must be pre-titrated. |
| NGS Library Prep Kit | For amplifying and barcoding sgRNA sequences from genomic DNA prior to sequencing. | Kits with high-fidelity polymerase (e.g., NEBNext) to minimize PCR bias. |
| sgRNA Read Alignment Pipeline | Software to demultiplex, quality-filter, and count sgRNA reads from FASTQ files. | MAGeCK FLUTE, CRISPResso2, or custom Python/R scripts. |
| Normalization & Analysis Tool | Statistical packages to normalize counts, calculate gene scores, and identify hits. | MAGeCK (RRA, MLE), BAGEL2 (Bayesian), PinAPL-Py (for plate screens). |
Conclusion
Effective data normalization is not merely a preprocessing step but a fundamental determinant of success in CRISPR screening. As outlined, a deep foundational understanding enables the selection of appropriate methodologies, while robust troubleshooting ensures data integrity. The comparative validation of methods highlights that there is no universal solution; the optimal strategy depends on screen design, biological context, and desired outcomes. Looking ahead, the integration of machine learning for adaptive normalization and the development of standardized benchmarks will be crucial as CRISPR screens grow in scale and complexity, moving towards more predictive models in therapeutic discovery and functional genomics. Mastering these normalization techniques is essential for transforming raw sequencing data into reliable, actionable biological knowledge.