CRISPRko vs CRISPRi vs CRISPRa: Choosing the Right Tool for Gene Function Analysis
This article provides a comprehensive guide for researchers and drug development professionals on the core CRISPR technologies for gene perturbation: knockout (CRISPRko), interference (CRISPRi), and activation (CRISPRa).
CRISPRko vs CRISPRi vs CRISPRa: Choosing the Right Tool for Gene Function Analysis
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on the core CRISPR technologies for gene perturbation: knockout (CRISPRko), interference (CRISPRi), and activation (CRISPRa). We cover foundational principles, methodological workflows, optimization strategies, and comparative validation to empower scientists in selecting the optimal approach for their functional genomics, screening, and therapeutic target discovery projects.
CRISPR Toolkit Basics: Decoding the Mechanisms of Ko, i, and a
This technical guide explores the evolution of CRISPR-Cas systems from simple endonucleases for gene knockout (CRISPRko) to sophisticated platforms for transcriptional modulation (CRISPRi/a) and precise epigenetic editing. The discussion is framed within the critical comparative analysis of CRISPRko, CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa), which are foundational tools for functional genomics and therapeutic development.
Core Mechanisms and Comparative Analysis
Fundamental Nuclease: CRISPRko
CRISPRko utilizes the endonuclease activity of Cas9 or Cas12 to create double-strand breaks (DSBs), leading to frameshift mutations and gene knockout via the error-prone non-homologous end joining (NHEJ) pathway.
Transcriptional Modulation: CRISPRi & CRISPRa
CRISPRi employs a catalytically "dead" Cas9 (dCas9) fused to transcriptional repressor domains (e.g., KRAB) to sterically block RNA polymerase or recruit chromatin-condensing machinery. CRISPRa utilizes dCas9 fused to transcriptional activator domains (e.g., VP64, p65AD) to recruit co-activators and open chromatin, upregulating target gene expression.
Table 1: Core Comparison of CRISPRko, CRISPRi, and CRISPRa
| Feature | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Cas Protein | Wild-type Cas9/Cas12 | dCas9 (H840A, D10A) | dCas9 (H840A, D10A) |
| Primary Function | Permanent gene disruption | Reversible gene silencing | Gene upregulation |
| Key Fusion Domains | None (nuclease) | KRAB, SID, Mxi1 | VP64, p65AD, Rta, SunTag |
| Mechanism | DSB → NHEJ/Indel | Steric hindrance & chromatin repression | Recruitment of transcriptional machinery |
| Efficiency (Typical) | 40-80% indel formation | 50-90% repression (varies by gene) | 2-20x activation (varies by gene) |
| Reversibility | No | Yes | Yes |
| Primary Applications | Essential gene studies, loss-of-function | Tuning gene expression, essential gene study | Gain-of-function, gene overexpression |
| Common Delivery | Plasmid, RNP | Plasmid, Lentivirus | Plasmid, Lentivirus |
Epigenetic Modulators
The dCas9 scaffold is further fused to epigenetic writer/eraser enzymes (e.g., DNMT3A for DNA methylation, TET1 for demethylation, p300 for histone acetylation) to create lasting epigenetic marks without altering the DNA sequence.
Table 2: Quantitative Performance Metrics of Epigenetic Editors
| Epigenetic Editor | Catalytic Domain | Target Modification | Typical Efficiency (vs. Control) | Persistence (Duration) | Key Readout |
|---|---|---|---|---|---|
| dCas9-DNMT3A | DNMT3A | CpG Methylation | 20-50% increase in mCpG | Weeks to months (mitotic) | Bisulfite sequencing |
| dCas9-TET1 | TET1 | CpG Demethylation | 30-70% decrease in mCpG | Weeks to months (mitotic) | Bisulfite sequencing |
| dCas9-p300 | p300 core | H3K27ac Acetylation | 5-20 fold increase in acetylation | Days to weeks | ChIP-qPCR/Seq |
| dCas9-LSD1 | LSD1 | H3K4me1/2 Demethylation | 50-80% reduction in methylation | Days to weeks | ChIP-qPCR/Seq |
Experimental Protocols
Protocol: CRISPRko Screen with NGS Readout
Objective: Perform a genome-wide loss-of-function screen.
- Library Design: Use a pooled lentiviral sgRNA library (e.g., Brunello, 4 sgRNAs/gene).
- Cell Transduction: Infect target cells at an MOI of ~0.3 to ensure single integration. Select with puromycin (1-3 µg/mL) for 5-7 days.
- Phenotype Induction: Split cells into experimental (e.g., drug treatment) and control arms. Maintain for 14-21 cell doublings.
- Genomic DNA Extraction: Harvest ≥1e7 cells per arm. Extract gDNA using a large-scale kit (e.g., Qiagen Blood & Cell Culture DNA Maxi Kit).
- sgRNA Amplification: Perform a two-step PCR (Step 1: amplify sgRNA region; Step 2: add Illumina adapters and sample indexes). Use 10 µg gDNA per 100 µL PCR reaction.
- Sequencing & Analysis: Pool PCR products and sequence on an Illumina NextSeq (75 bp single-end). Align reads to the library and analyze sgRNA depletion/enrichment using MAGeCK or BAGEL2.
Protocol: Targeted Transcriptional Activation (CRISPRa)
Objective: Activate a specific endogenous gene.
- System Design: Use a dCas9-VP64-p65AD (VPR) fusion construct and a synergistic activator sgRNA (sa-sgRNA) scaffold.
- Delivery: Co-transfect HEK293T cells with dCas9-VPR plasmid (500 ng) and sa-sgRNA plasmid (250 ng) per well of a 24-well plate using PEI transfection reagent.
- Harvest: 48-72 hours post-transfection, harvest cells.
- Validation:
- qRT-PCR: Isolate RNA, synthesize cDNA, perform qPCR with target-specific primers. Express as fold-change over non-targeting sgRNA control (2^-ΔΔCt method).
- Western Blot: Confirm increased protein expression.
Protocol: DNA Methylation Editing with dCas9-DNMT3A
Objective: Induce de novo methylation at a specific promoter.
- Design: Clone dCas9-DNMT3A-3L (with added nuclear localization signals) into a lentiviral vector. Design sgRNAs targeting the CpG island of the promoter.
- Stable Cell Line Generation: Produce lentivirus and transduce target cells. Select with blasticidin (5-10 µg/mL) for 10 days to create a polyclonal stable line expressing the editor.
- Methylation Editing: Transfect stable cells with sgRNA expression plasmid. Maintain cells for 7-14 days to allow methylation accumulation.
- Analysis:
- Bisulfite Sequencing: Treat genomic DNA with sodium bisulfite. Amplify target region by PCR, clone products, and sequence 10-20 clones to determine percentage methylation per CpG site.
- Phenotypic Assay: Measure downstream gene expression (qRT-PCR).
Signaling Pathways and Workflows
Diagram Title: Evolution from CRISPRko to Epigenetic Editing
Diagram Title: Decision Workflow for CRISPRko, i, a, and Epigenetic Editing
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-Based Experiments
| Reagent / Material | Supplier Examples | Function & Brief Explanation |
|---|---|---|
| High-Efficiency dCas9 Fusion Plasmids | Addgene, Thermo Fisher | Source of pre-cloned, validated dCas9-effector constructs (KRAB, VPR, p300, DNMT3A) for reliable expression. |
| Validated sgRNA Cloning Libraries | Horizon (Dharmacon), Sigma | Pre-designed, sequence-verified pooled or arrayed sgRNA libraries for specific genomes and applications. |
| Lentiviral Packaging Mix (psPAX2, pMD2.G) | Addgene, Invitrogen | Essential plasmids for producing safe, high-titer lentivirus to deliver CRISPR components stably. |
| Cas9/dCas9 Recombinant Protein | IDT, Thermo Fisher | Purified protein for rapid, transient RNP (ribonucleoprotein) delivery, reducing off-target effects. |
| Lipofectamine CRISPRMAX / Cas9 Plus | Thermo Fisher | Optimized lipid nanoparticles for high-efficiency, low-toxicity delivery of CRISPR RNPs or plasmids. |
| Next-Generation Sequencing Kits for CRISPR Screens | Illumina, Qiagen | Kits for amplifying and preparing sgRNA libraries from genomic DNA for deep sequencing analysis. |
| Methylation-Specific PCR (MSP) or Bisulfite Conversion Kits | Qiagen, Zymo Research | Essential for analyzing DNA methylation outcomes following epigenetic editing (e.g., with dCas9-DNMT3A). |
| Chromatin Immunoprecipitation (ChIP) Kits | Abcam, Cell Signaling Tech | Required for validating histone modification changes (e.g., H3K27ac) after CRISPRa or epigenetic editing. |
| Validated Antibodies for Epigenetic Marks | Active Motif, Abcam | Antibodies specific to modifications (H3K4me3, H3K9me3, H3K27ac) to confirm on-target epigenetic editing. |
| Cell Viability/Proliferation Assay Kits | Promega (CellTiter-Glo) | Quantitative readouts for functional consequences of CRISPRko/i/a screens or edits. |
1. Introduction Within the landscape of programmable gene regulation tools, CRISPRko (CRISPR knockout) stands as the definitive method for achieving permanent, complete loss-of-function. Its mechanism is distinct from its reversible counterparts, CRISPR interference (CRISPRi) for gene silencing and CRISPR activation (CRISPRa) for gene upregulation. This technical guide details the core molecular and cellular processes by which CRISPRko-induced double-strand breaks (DSBs) lead to irreversible gene knockout, providing essential context for researchers selecting the appropriate modality for functional genomics and therapeutic development.
2. Core Mechanism: From DSB to Frameshift Mutation CRISPRko utilizes a catalytically active Cas nuclease (commonly SpCas9) guided by a single-guide RNA (sgRNA) to create a targeted DSB within an early exon of the gene of interest. The cell's primary repair pathways then dictate the outcome.
Table 1: Comparison of Major DNA Repair Pathways Engaged After a CRISPRko DSB
| Pathway | Key Enzymes/Factors | Fidelity | Typical Outcome for CRISPRko | Frequency at CRISPR Cut Sites* |
|---|---|---|---|---|
| Non-Homologous End Joining (NHEJ) | DNA-PKcs, Ku70/80, XLF, XRCC4, DNA Ligase IV | Error-prone | Small insertions or deletions (indels). Leads to frameshifts and premature stop codons. | High (~60-80%) |
| Microhomology-Mediated End Joining (MMEJ) | PARP1, CtIP, MRE11, DNA Ligase 1/3 | Error-prone | Larger deletions flanking microhomology sequences. Leads to exon loss or major gene disruption. | Moderate (~10-20%) |
| Homology-Directed Repair (HDR) | BRCA1, BRCA2, RAD51, Exogenous DNA template | High-fidelity | Precise, templated repair. Can be co-opted for knock-in, but is rare in non-dividing cells. | Low (<5-10% in dividing cells without selection) |
*Frequencies are approximate and highly dependent on cell type, cell cycle stage, and genomic context.
3. Detailed Experimental Protocol: A Standard CRISPRko Workflow Protocol: Generating a Clonal Knockout Cell Line Using SpCas9
- sgRNA Design & Cloning: Design 2-3 sgRNAs targeting early exons of the target gene. Clone oligonucleotides into a Cas9/sgRNA expression vector (e.g., lentiCRISPRv2, pX458).
- Delivery: Transfect or transduce the target cell line (e.g., HEK293T, iPSCs) with the sgRNA/Cas9 construct.
- Transient Enrichment (Optional): If using a plasmid with a fluorescent marker (e.g., GFP), use FACS to sort transfected cells 48-72 hours post-delivery.
- Clonal Isolation: Plate cells at low density to derive single-cell clones. Allow 1-3 weeks for colony formation.
- Genotyping:
- Genomic DNA Extraction: Harvest clonal cells and extract gDNA.
- PCR Amplification: Amplify the targeted genomic region (~500-700bp).
- Analysis: Use Sanger sequencing followed by TIDE or ICE analysis to quantify editing efficiency and identify frameshift mutations, or perform Next-Generation Sequencing (NGS) of the PCR amplicon for deep characterization.
- Phenotypic Validation: Confirm knockout via Western blot (protein loss) and/or a functional assay specific to the gene.
4. The DSB Repair Pathway Decision Logic
5. The Scientist's Toolkit: Essential Research Reagents for CRISPRko
Table 2: Key Research Reagent Solutions for CRISPRko Experiments
| Reagent / Material | Function & Importance | Example Products/Vendors |
|---|---|---|
| Cas9 Expression Vector | Delivers the Cas9 nuclease. May be all-in-one with sgRNA scaffold. | lentiCRISPRv2 (Addgene), pSpCas9(BB) (Addgene), TrueCut Cas9 Protein (Thermo Fisher) |
| sgRNA Cloning Vector | Backbone for synthesizing and expressing target-specific sgRNA. | pGL3-U6-sgRNA (Addgene), commercial sgRNA synthesis kits |
| NHEJ Inhibitor (Optional) | Shifts repair balance towards HDR; used to test pathway dependence. | SCR7, Nu7026 |
| HDR Donor Template | Single-stranded oligodeoxynucleotide (ssODN) or double-stranded DNA for knock-in controls. | Ultramer DNA Oligos (IDT), gBlocks (IDT) |
| Genotyping Primers | Flank the target site for PCR amplification prior to sequencing. | Custom DNA Oligos (any supplier) |
| Indel Analysis Software | Quantifies editing efficiency and predicts frameshifts from sequencing data. | TIDE, ICE (Synthego), CRISPResso2 |
| Positive Control sgRNA | Targets a housekeeping gene with known phenotype (e.g., AAVS1 safe harbor). | Validated controls available from supplier libraries (e.g., Sigma, Origene) |
| Transfection/Transduction Reagent | Enables delivery of CRISPR constructs into target cells. | Lipofectamine CRISPRMAX (Thermo Fisher), Polybrene (for lentiviral delivery), Neon Transfection System |
6. Advanced Considerations & Protocol: Validating Knockout Specificity
Protocol: Off-Target Analysis by GUIDE-seq or CIRCLE-seq
- GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by sequencing):
- Co-deliver Cas9-sgRNA RNP with a blunt-ended, double-stranded oligonucleotide ("tag") into cells.
- The tag integrates into DSB sites via NHEJ.
- Extract genomic DNA, shear, and perform PCR amplification using a tag-specific primer.
- Sequence amplicons via NGS and map reads to the genome to identify all integration sites (on- and off-target).
- CIRCLE-seq (Circularization for In Vitro Reporting of Cleavage Effects by sequencing):
- Isolate genomic DNA and shear it.
- Circulate the fragments and digest with a nuclease to linearize only circles containing a Cas9 cut site.
- Adapter-ligate and sequence the linearized fragments via NGS.
- Bioinformatics analysis identifies sequences with high similarity to the sgRNA that were cleaved in vitro.
7. Conclusion: Strategic Positioning of CRISPRko CRISPRko's reliance on error-prone NHEJ/MMEJ repair to generate disruptive indels makes it the unrivaled choice for complete, stable gene ablation. This contrasts with CRISPRi's reversible suppression via dCas9-KRAB-mediated heterochromatin formation and CRISPRa's targeted gene upregulation via dCas9-VPR transcriptional activation. The selection among these tools hinges on the experimental requirement for permanence, reversibility, or gain-of-function, with CRISPRko remaining the cornerstone for definitive loss-of-function studies in drug target validation and functional genomics.
CRISPR interference (CRISPRi) is a refined technique for sequence-specific gene silencing without introducing double-strand DNA breaks. It is one of three principal modalities for CRISPR-based transcriptional regulation, distinct from CRISPR knockout (CRISPRko) and CRISPR activation (CRISPRa). Understanding their differences is crucial for selecting the appropriate research tool.
The core distinction lies in the endonuclease activity of the Cas protein and the recruitment of effector domains. CRISPRko utilizes a catalytically active Cas9 (or Cas12) to create irreversible double-strand breaks, leading to frameshift mutations and gene knockout. In contrast, CRISPRi employs a catalytically dead Cas9 (dCas9) fused to a transcriptional repressor domain (e.g., KRAB). This complex binds to the target DNA sequence, typically within the promoter or early coding region, and silences transcription by sterically hindering RNA polymerase or recruiting chromatin-modifying complexes. CRISPRa also uses dCas9, but fused to transcriptional activator domains (e.g., VPR, p65AD), to upregulate gene expression.
The choice between these platforms hinges on the experimental goal: permanent loss-of-function (CRISPRko), reversible and titratable downregulation (CRISPRi), or gain-of-function studies (CRISPRa). CRISPRi offers significant advantages for studying essential genes, creating hypomorphic alleles, and conducting functional genomics screens with minimal off-target phenotypic consequences.
Core Mechanism and Signaling Pathways
CRISPRi functions through the targeted recruitment of repressive chromatin machinery to specific genomic loci. The primary components are a single-guide RNA (sgRNA) and a dCas9 protein fused to a repressor domain. The most common repressor is the Kruppel-associated box (KRAB) domain from human KOX1.
Mechanism Diagram:
Quantitative Comparison: CRISPRko vs. CRISPRi vs. CRISPRa
Table 1: Core Functional Comparison of CRISPR Modulation Platforms
| Feature | CRISPR Knockout (CRISPRko) | CRISPR Interference (CRISPRi) | CRISPR Activation (CRISPRa) |
|---|---|---|---|
| Cas Protein | Wild-type SpCas9 (or other nucleases) | Catalytically dead Cas9 (dCas9) | Catalytically dead Cas9 (dCas9) |
| Core Effector | Nuclease domains | Repressor domain (e.g., KRAB) | Activator domain (e.g., VPR, p65AD) |
| DNA Cleavage | Yes, creates DSBs | No | No |
| Primary Outcome | Irreversible frameshift mutations & indels | Reversible transcriptional repression | Transcriptional activation |
| Reversibility | No (permanent) | Yes (transient upon complex removal) | Yes (transient upon complex removal) |
| Typical Efficiency | High (70-90% indel rate) | High (70-95% repression) | Moderate-High (5-50x activation) |
| Key Applications | Complete gene loss-of-function, screening | Essential gene studies, tunable knockdown, functional screening | Gain-of-function, genetic suppression, overexpression screens |
| Off-Target Concerns | DSB-related toxicity, translocations | Minimal (no DSBs), potential binding site competition | Minimal (no DSBs), potential binding site competition |
Table 2: Performance Metrics from Recent Studies (2023-2024)
| Parameter | CRISPRi (dCas9-KRAB) | Source / Notes |
|---|---|---|
| Max Repression Efficiency | Up to 99.9% (10-fold reduction) | Varies by gene and sgRNA design; typical range 80-95%. |
| Onset of Repression | 24-48 hours post-transfection | Time to achieve steady-state mRNA reduction. |
| Duration of Effect | Stable with continuous expression; reversible within 3-7 days upon loss of dCas9/sgRNA. | Dependent on cell division and complex dilution. |
| Optimal Targeting Region | -50 to +300 bp relative to TSS. | Most effective within the promoter or early exon. |
| Multiplexing Capacity | Demonstrated with ≥7 genes simultaneously. | Limited by delivery vector capacity and competition. |
Detailed Experimental Protocol for a CRISPRi Knockdown Experiment
A. sgRNA Design and Cloning
- Design: Select 3-5 sgRNAs targeting the non-template strand of the gene promoter region, -50 to +300 bp from the transcription start site (TSS). Use validated algorithms (e.g., CRISPick, CHOPCHOP) to minimize off-target effects.
- Oligos: Synthesize oligonucleotides: Forward: 5'-CACCG[N20] -3', Reverse: 5'-AAAC[N20]C-3'.
- Cloning into Lentiviral Vector:
- Digest the lentiviral CRISPRi plasmid (e.g., pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro) with BsmBI.
- Gel-purify the linearized vector.
- Anneal the oligos (95°C for 5 min, ramp down to 25°C) to form a duplex with BsmBI-compatible overhangs.
- Ligate the duplex into the digested vector using T4 DNA ligase.
- Transform into competent E. coli, plate on ampicillin, and screen colonies by Sanger sequencing using a U6 promoter primer.
B. Lentivirus Production and Transduction
- Day 1: Seed HEK293T cells in a 6-well plate at 70% confluence.
- Day 2: Transfect using a suitable reagent (e.g., PEI). For one well, mix:
- 1 µg of sgRNA expression plasmid
- 0.9 µg of psPAX2 (packaging plasmid)
- 0.1 µg of pMD2.G (VSV-G envelope plasmid)
- in 100 µL serum-free DMEM.
- Add 6 µL of 1 mg/mL PEI, vortex, incubate 15 min, and add dropwise to cells.
- Day 3 & 4: Replace medium with fresh complete medium.
- Day 5: Harvest viral supernatant, filter through a 0.45 µm PVDF filter.
- Transduction: Incubate target cells with viral supernatant plus polybrene (8 µg/mL) for 24 hours. Replace with fresh medium.
C. Selection and Validation
- Selection: 48 hours post-transduction, begin selection with puromycin (concentration determined by kill curve, typically 1-5 µg/mL). Maintain selection for 5-7 days.
- Validation of Knockdown:
- qRT-PCR (mRNA level): Extract total RNA 5-7 days post-selection. Perform reverse transcription and qPCR with primers in the target gene's coding sequence, normalizing to housekeeping genes (e.g., GAPDH, ACTB).
- Western Blot (Protein level): Harvest protein lysates 7-10 days post-selection. Probe for the target protein and a loading control (e.g., β-Actin).
- Functional Assay: Perform a phenotype-specific assay relevant to the gene's function.
Workflow Diagram:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for CRISPRi Experiments
| Reagent / Material | Function & Explanation | Example Product/Catalog # (Representative) |
|---|---|---|
| dCas9-KRAB Expression Vector | Stable expression of the dead Cas9 fused to the KRAB repression domain. Essential for CRISPRi activity. | pLV hU6-sgRNA hUbC-dCas9-KRAB-Puro (Addgene #71236) |
| sgRNA Cloning Backbone | Lentiviral vector with a U6 promoter for sgRNA expression and a resistance marker. | lentiGuide-Puro (Addgene #52963) |
| Lentiviral Packaging Plasmids | psPAX2 (gag/pol) and pMD2.G (VSV-G env) for producing replication-incompetent lentivirus. | psPAX2 (Addgene #12260), pMD2.G (Addgene #12259) |
| Transfection Reagent | For delivering plasmids into HEK293T cells during virus production. High efficiency required. | Polyethylenimine (PEI) Max, Lipofectamine 3000 |
| Polybrene (Hexadimethrine bromide) | A cationic polymer that enhances viral transduction efficiency by neutralizing charge repulsion. | Typically used at 4-8 µg/mL. |
| Selection Antibiotic | To select for cells successfully expressing the CRISPRi construct. Must match the vector's resistance marker. | Puromycin dihydrochloride (for puromycin resistance). |
| Validated sgRNA Libraries | Pre-designed, arrayed or pooled libraries targeting specific gene families or genome-wide for screens. | Human CRISPRi v2 (Brunello) library (Addgene #83978) |
| dCas9 Antibody | For validating dCas9-KRAB fusion protein expression via Western blot. | Anti-Cas9 antibody (7A9-3A3, Cell Signaling #14697) |
| qRT-PCR Reagents | For quantifying mRNA knockdown efficiency. Includes reverse transcriptase, SYBR Green master mix, gene-specific primers. | iTaq Universal SYBR Green Supermix, High-Capacity cDNA Reverse Transcription Kit |
CRISPR activation (CRISPRa) represents a powerful gain-of-function approach within the CRISPR toolbox. To understand its unique position, it is essential to contrast it with the other primary modalities: CRISPR knockout (CRISPRko) and CRISPR interference (CRISPRi). This whitepaper provides an in-depth technical guide to CRISPRa, framed within the broader thesis of selecting the appropriate CRISPR-based perturbation method for functional genomics and therapeutic development.
Core Functional Thesis:
- CRISPRko (Knockout): Utilizes Cas9 nuclease to create double-strand breaks, leading to frameshift mutations and permanent gene disruption via non-homologous end joining (NHEJ). It is the standard for loss-of-function studies.
- CRISPRi (Interference): Employs a catalytically dead Cas9 (dCas9) fused to transcriptional repressors (e.g., KRAB, SID4x) to block transcription initiation or elongation, resulting in reversible gene silencing.
- CRISPRa (Activation): Leverages dCas9 fused to transcriptional activators (e.g., VPR, SAM) to recruit the cellular transcription machinery to specific promoter or enhancer regions, enabling precise and robust gene upregulation.
The choice between these systems depends on the biological question: permanent loss (ko), reversible suppression (i), or controlled overexpression (a).
Core CRISPRa Architectures: Mechanisms and Components
CRISPRa systems are built upon a dCas9 scaffold, which provides programmable DNA binding without cleavage. The key innovation is the fusion or recruitment of transcriptional activation domains.
Primary Activation Systems
1. dCas9-VP64: The pioneering system, where dCas9 is fused to a tetramer of the VP16 activation domain (VP64). It offers modest activation and often requires multiple guide RNAs (gRNAs) targeting the same promoter for synergistic effect.
2. dCas9-SunTag: A recruiting platform where dCas9 is fused to an array of peptide epitopes (SunTag). Co-expressed single-chain variable fragment (scFv) antibodies, fused to VP64, bind to the SunTag, resulting in the recruitment of multiple activators to a single dCas9 molecule, enhancing potency.
3. dCas9-VPR: A direct fusion of dCas9 to a tripartite activator: VP64, p65, and Rta (VPR). This chimeric activator provides significantly stronger transcriptional upregulation than VP64 alone.
4. SAM (Synergistic Activation Mediator): A sophisticated three-component system: * dCas9-VP64: Serves as the DNA-targeting base. * MS2-p65-HSF1: A modified gRNA with MS2 RNA aptamers recruits the MS2 coat protein (MCP) fused to the p65-HSF1 activation domains. This system creates a synergistic recruitment of multiple distinct activators, leading to very high levels of gene activation.
Quantitative Comparison of Activation Systems
Table 1: Performance Metrics of Common CRISPRa Systems (Representative Data)
| System | Relative Activation Fold-Change (Range) | Typical gRNA Targeting Region | Multiplexing Capacity | Key Advantage |
|---|---|---|---|---|
| dCas9-VP64 | 2x - 10x | -200 to -50 bp from TSS | Low | Simple, minimal size |
| dCas9-SunTag | 10x - 100x | -200 to -50 bp from TSS | Medium | Amplified recruitment |
| dCas9-VPR | 50x - 500x | -200 to +1 bp from TSS | Low | High potency, single construct |
| SAM | 100x - 1000x+ | -200 to +1 bp from TSS | High | Very high-level activation, modular |
Detailed Experimental Protocol for CRISPRa
Protocol: Setting Up a CRISPRa Experiment Using the SAM System
Objective: To achieve robust, inducible upregulation of a target gene in a human cell line (e.g., HEK293T).
I. Design and Cloning of gRNA Expression Construct
- gRNA Design:
- Identify the target promoter. For SAM, design gRNAs within -200 to +1 bp relative to the Transcription Start Site (TSS).
- Use established algorithms (e.g., CRISPick, CHOPCHOP) to predict high-activity gRNAs and minimize off-target effects.
- Incorporate two MS2 RNA aptamer loops into the tracrRNA sequence of the gRNA scaffold (using a plasmid backbone like lenti-MS2-P65-HSF1-Hygro from Addgene #89308).
- Cloning: Clone the synthesized target-specific 20nt spacer sequence into the BsmBI site of the chosen gRNA expression vector via Golden Gate assembly.
II. Lentiviral Production and Cell Line Engineering
- Co-transfection for Virus Production:
- In HEK293T packaging cells, co-transfect three plasmids using PEI or a commercial reagent: a. SAM gRNA plasmid (from step I). b. dCas9-VP64_Blast plasmid (Addgene #61425). c. Viral packaging plasmids (psPAX2 and pMD2.G).
- Harvest lentiviral supernatant at 48 and 72 hours post-transfection.
- Transduction and Selection:
- Transduce target cells with filtered viral supernatant in the presence of polybrene (8 µg/mL).
- 48 hours post-transduction, begin selection with appropriate antibiotics (e.g., Blasticidin for dCas9-VP64, Hygromycin for the MS2-gRNA construct). Maintain selection for 5-7 days to establish a polyclonal population.
III. Validation and Analysis
- qRT-PCR Analysis:
- Isolate total RNA from selected cells using TRIzol.
- Synthesize cDNA using a reverse transcription kit with random hexamers.
- Perform quantitative PCR (qPCR) using primers specific to the target gene's coding sequence. Normalize expression to housekeeping genes (e.g., GAPDH, ACTB).
- Calculate fold-change using the 2^(-ΔΔCt) method compared to a non-targeting gRNA control.
- Functional Assay: Perform a phenotype-specific assay (e.g., ELISA for secreted protein, flow cytometry for a surface marker, cell proliferation assay) to confirm the functional consequence of gene upregulation.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for CRISPRa
| Reagent / Material | Function / Purpose | Example Source / Identifier |
|---|---|---|
| dCas9-VPR Plasmid | All-in-one vector for direct fusion CRISPRa system. | Addgene #63798 |
| SAM System Plasmids (3-Part) | Separate vectors for dCas9-VP64, MS2-gRNA, and MCP-p65-HSF1. | Addgene #61425, #89308 |
| Lentiviral Packaging Mix | Produces replication-incompetent lentivirus for stable cell line generation. | psPAX2 (Addgene #12260), pMD2.G (Addgene #12259) |
| Validated Non-Targeting gRNA | Critical negative control for gRNA-specific effects. | Addgene #109381 (hU6-MSH-gRNA) |
| T7 Endonuclease I / Surveyor Kit | Assesses potential off-target cleavage (more relevant for CRISPRko). | NEB #M0302S |
| CRISPRa gRNA Design Tool | Web-based platform for predicting high-efficiency gRNAs. | CRISPick (broadinstitute.org) |
| Polybrene (Hexadimethrine Bromide) | Enhances viral transduction efficiency in cell culture. | Sigma-Aldrich #H9268 |
| Blasticidin S HCl | Selective antibiotic for dCas9-VP64/blast plasmid. | Thermo Fisher #A1113903 |
| Hygromycin B | Selective antibiotic for MS2-gRNA/hygro plasmid. | Thermo Fisher #10687010 |
Comparative Analysis: CRISPRko vs. CRISPRi vs. CRISPRa
Table 3: Strategic Comparison of Core CRISPR Perturbation Modalities
| Feature | CRISPRko (Knockout) | CRISPRi (Interference) | CRISPRa (Activation) |
|---|---|---|---|
| Cas Protein | Cas9 nuclease (WT) | dCas9 fused to repressor (e.g., KRAB) | dCas9 fused to activator (e.g., VPR, SAM) |
| Primary Effect | Permanent DNA disruption, protein loss | Reversible transcriptional repression | Transcriptional upregulation |
| Kinetics | Permanent after editing; effect depends on protein turnover | Rapid (hours), reversible upon removal of dCas9-i | Rapid (hours), tunable by expression level |
| gRNA Target | Early exons (to disrupt ORF) | Promoter or TSS (to block Pol II) | Promoter or enhancer (to recruit Pol II) |
| Key Application | Essential gene studies, tumor suppressor validation | Essential gene studies (non-lethal), pathway dampening | Gain-of-function, gene dosage studies, differentiation |
| Main Advantage | Complete, permanent loss-of-function | Reversible, fewer off-target mutations than KO | Precise, tunable gain-of-function |
| Main Limitation | Off-target indels, lethal for essential genes | "Leaky" repression, incomplete knockdown | Context-dependent efficiency, potential for overexpression artifacts |
Advanced Considerations and Future Directions
- Epigenetic Modulation: Next-generation CRISPRa systems are incorporating epigenetic writers (e.g., p300, TET1) to alter chromatin marks (H3K27ac, DNA methylation) for more stable or potent activation.
- Spatiotemporal Control: Fusion of dCas9-activators to inducible degrons or light-sensitive domains (Optogenetics) allows precise control over the timing and magnitude of activation.
- Therapeutic Applications: CRISPRa is being explored for diseases caused by haploinsufficiency (e.g., certain forms of Parkinson's, metabolic disorders) where upregulating the functional copy of a gene could restore normal cellular function.
- Multiplexed Activation: Libraries of gRNAs targeting multiple genes or pathways enable genome-wide or pathway-specific activation screens to identify genes that confer therapeutic resistance or drive cell fate changes.
In conclusion, CRISPRa fills the critical gain-of-function niche within the CRISPR ecosystem. Its strategic deployment, as contrasted with CRISPRko and CRISPRi, enables researchers to interrogate gene function, model disease, and develop novel therapeutic strategies with unprecedented precision and power.
CRISPR-based transcriptional modulation has revolutionized functional genomics by enabling precise control over gene expression without altering the underlying DNA sequence. This field stratifies into three core methodologies: CRISPR knockout (CRISPRko), which uses wild-type Cas9 to create double-strand breaks for gene disruption; CRISPR interference (CRISPRi), which employs a catalytically dead Cas9 (dCas9) fused to repressive domains like KRAB to silence gene expression; and CRISPR activation (CRISPRa), which utilizes dCas9 fused to transcriptional activators like VPR to upregulate gene expression. This whitepaper provides an in-depth technical analysis of the core protein variants—dCas9, dCas9-KRAB, and dCas9-VPR—that are fundamental to the CRISPRi and CRISPRa modalities, contrasting them with the nuclease-dependent CRISPRko approach.
Core Protein Variants: Structure, Function, and Quantitative Performance
dCas9: The Foundational Scaffold
The dCas9 variant is generated through point mutations (commonly D10A and H840A for Streptococcus pyogenes Cas9) that abolish its endonuclease activity while retaining high-affinity, guide RNA-programmed DNA binding. This creates a versatile DNA-targeting platform that sterically blocks transcription initiation or elongation when bound within a promoter or coding region, leading to modest transcriptional repression (typically 2-5 fold).
dCas9-KRAB: The Potent Repressor
dCas9-KRAB is created by fusing the Krüppel-associated box (KRAB) domain from human KOX1 to the C-terminus of dCas9. The KRAB domain recruits endogenous repressive complexes, including heterochromatin-forming factors like HP1 and histone methyltransferases (e.g., SETDB1), leading to histone H3 lysine 9 trimethylation (H3K9me3) and stable, heritable gene silencing. This fusion dramatically enhances repression efficacy over dCas9 alone.
dCas9-VPR: The Robust Activator
dCas9-VPR is a tripartite activator fusion, where dCas9 is linked to a tandem array of three potent activation domains: VP64, p65, and Rta (VPR). This combination synergistically recruits coactivators and the general transcription machinery, leading to strong transcriptional upregulation. It is significantly more potent than earlier single-domain activators like dCas9-VP64.
Table 1: Quantitative Performance Comparison of Core dCas9 Variants
| Variant | Core Function | Typical Fold-Change (vs. Control) | Key Effector Domain(s) | Primary Chromatin Modification | Optimal Targeting Region |
|---|---|---|---|---|---|
| dCas9 | Steric Blockade / Mild Repression | 0.2 - 0.5x (Repression) | None (Steric Hindrance) | N/A | -50 to +300 bp from TSS |
| dCas9-KRAB | Epigenetic Silencing | 0.01 - 0.1x (Repression) | KRAB domain | H3K9me3 | -50 to +1 bp from TSS |
| dCas9-VPR | Transcriptional Activation | 10 - 1000x (Activation) | VP64, p65, Rta | H3K27ac, H3K4me3 | -400 to -50 bp from TSS |
Table 2: Comparative Overview: CRISPRko vs. CRISPRi vs. CRISPRa
| Feature | CRISPRko | CRISPRi (dCas9-KRAB) | CRISPRa (dCas9-VPR) |
|---|---|---|---|
| Cas Protein | Wild-type Cas9 | dCas9-KRAB | dCas9-VPR |
| DNA Cleavage | Yes | No | No |
| Primary Outcome | Indel formation, gene knockout | Reversible transcriptional repression | Transcriptional activation |
| Mechanism | NHEJ/MMEJ-mediated repair errors | Chromatin compaction & silencing | Recruitment of transcriptional machinery |
| Efficacy | High (near-complete protein loss) | High (90-99% knockdown) | Variable (often 10-1000x upregulation) |
| Reversibility | Permanent | Reversible (epigenetic) | Reversible (epigenetic) |
| Off-Target Concerns | DNA sequence alterations | Transcriptional/Epigenetic only | Transcriptional/Epigenetic only |
| Typical Application | Essential gene studies, loss-of-function screens | Functional knockdown, synthetic circuits, disease modeling | Gain-of-function screens, gene therapy, differentiation |
Experimental Protocols for Key Applications
Protocol: Genome-Scale CRISPRi/KRAB Knockdown Screen
Objective: Identify essential genes in a cell line using a pooled dCas9-KRAB sgRNA library. Materials: See "Scientist's Toolkit" (Section 5). Method:
- Stable Cell Line Generation: Lentivirally transduce target cells with a dCas9-KRAB expression construct. Select with appropriate antibiotics (e.g., blasticidin) for 7-10 days to create a polyclonal stable line.
- Library Transduction: Infect the dCas9-KRAB cells at low MOI (~0.3) with the lentiviral sgRNA library (e.g., Brunello CRISPRi library). Ensure library coverage of >500 cells per sgRNA. Select with puromycin for 7 days.
- Screening: Passage cells for 14-21 population doublings. Maintain sufficient cell numbers (>1000x library representation) at each passage to prevent bottlenecking.
- Genomic DNA Extraction & NGS: Harvest genomic DNA from the initial (T0) and final (Tfinal) cell populations using a large-scale gDNA kit. Amplify the integrated sgRNA sequences via PCR using indexed primers.
- Sequencing & Analysis: Perform deep sequencing (Illumina). Align reads to the library reference. Use statistical packages (e.g., MAGeCK) to compare sgRNA abundance between T0 and Tfinal, identifying significantly depleted sgRNAs and their target essential genes.
Protocol: Targeted Transcriptional Activation with dCas9-VPR
Objective: Activate a specific endogenous gene and measure mRNA output. Method:
- sgRNA Design: Design 3-5 sgRNAs targeting the region -400 to -50 bp upstream of the target gene's transcription start site (TSS).
- Co-transfection: Co-transfect HEK293T cells (or target cell line) with two plasmids: (1) a dCas9-VPR expression plasmid, and (2) a plasmid expressing the target-specific sgRNA. Use a fluorescent reporter or antibiotic resistance marker for normalization.
- Validation & Harvest: 48-72 hours post-transfection, harvest cells.
- qRT-PCR Analysis: Isolve total RNA, synthesize cDNA, and perform quantitative PCR (qPCR) with primers specific to the target gene and housekeeping controls (e.g., GAPDH, ACTB). Calculate fold-change using the ΔΔCt method relative to a non-targeting sgRNA control.
Pathway and Workflow Visualizations
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for dCas9-KRAB/i and dCas9-VPR/a Experiments
| Reagent / Solution | Function / Description | Example Supplier/Catalog |
|---|---|---|
| dCas9-KRAB Expression Plasmid | Constitutively expresses the dCas9-KRAB fusion protein. Often lentiviral and includes a selection marker (e.g., Blasticidin R). | Addgene #71236 (pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Blast) |
| dCas9-VPR Expression Plasmid | Constitutively expresses the dCas9-VPR fusion protein. Selection marker varies. | Addgene #63798 (pHAGE EF1α dCas9-VPR) |
| Lentiviral sgRNA Library | Pooled, barcoded collection of sgRNA expression constructs for genome-wide screens (e.g., Brunello, Dolcetto). | Broad Institute GPP (Brunello CRISPRi library) |
| Individual sgRNA Cloning Vectors | Backbone for cloning custom sgRNA sequences (e.g., using BsmBI sites). | Addgene #104990 (pU6-sgRNA EF1Alpha-puro-T2A-BFP) |
| Lentiviral Packaging Plasmids | psPAX2 and pMD2.G for producing lentiviral particles of dCas9 or sgRNA constructs. | Addgene #12260 & #12259 |
| Polybrene (Hexadimethrine Bromide) | A cationic polymer that enhances viral transduction efficiency. | Sigma-Aldrich H9268 |
| Puromycin Dihydrochloride | Antibiotic for selecting cells successfully transduced with sgRNA vectors containing a puromycin resistance gene. | Thermo Fisher Scientific A1113803 |
| Blasticidin S HCl | Antibiotic for selecting cells expressing dCas9 constructs with a blasticidin resistance marker. | Thermo Fisher Scientific A1113903 |
| Next-Generation Sequencing Kit | For preparing sequencing libraries from amplified sgRNA inserts (e.g., Illumina Nextera XT). | Illumina FC-131-1096 |
| MAGeCK Software | Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout; standard for analyzing screen NGS data. | Open Source (https://sourceforge.net/p/mageck/wiki/Home/) |
This technical guide details the core molecular components enabling precise genome perturbation. Within the broader thesis comparing CRISPRko (knockout), CRISPRi (interference), and CRISPRa (activation), these components are the levers that determine the mode, efficiency, and specificity of the genetic intervention. The choice of sgRNA, effector domain, and delivery system directly dictates whether a gene is permanently silenced, transiently repressed, or transcriptionally upregulated, forming the foundational toolkit for functional genomics and therapeutic development.
sgRNA Design: The Targeting Module
The single guide RNA (sgRNA) is the determinant of genomic specificity, composed of a CRISPR RNA (crRNA) spacer sequence and a scaffold.
Core Design Principles
- Spacer Sequence (20nt): Must be complementary to the DNA target immediately preceding a Protospacer Adjacent Motif (PAM). For S. pyogenes Cas9 (SpCas9), the PAM is 5'-NGG-3'.
- Specificity: Minimize off-targets by assessing genome-wide homology. Mismatches at the PAM-distal "seed" region (positions 1-12) are more disruptive to binding.
- On-target Efficiency: Influenced by local chromatin accessibility, DNA sequence composition (e.g., avoid poly-T tracts), and secondary RNA structure.
Quantitative Parameters for Design
Table 1: Key Quantitative Parameters for sgRNA Design (SpCas9)
| Parameter | Optimal Range/Characteristic | Impact on Experiment |
|---|---|---|
| GC Content | 40-60% | Higher stability and efficiency; extremes reduce performance. |
| Out-of-Frame Score | High (for KO) | Predicts likelihood of frameshift mutation in coding exons. |
| Specificity Score | >90 (tool-dependent) | Predicts off-target potential; higher is better. |
| Distance to TSS | -50 to +300 bp (for i/a) | Critical for CRISPRi/a efficiency relative to Transcription Start Site. |
Protocol: In silico sgRNA Design for CRISPRko/i/a
Objective: Design specific and efficient sgRNAs targeting a gene of interest (GOI).
- Sequence Retrieval: Obtain the genomic sequence of the GOI (including promoter/enhancer regions) from databases like Ensembl or UCSC Genome Browser.
- PAM Identification: Scan for all 5'-NGG-3' PAM sequences (for SpCas9) on both strands.
- Candidate Extraction: Extract the 20nt genomic sequence directly 5' to each PAM as the potential spacer.
- Efficiency Scoring: Input candidate spacers into predictive algorithms (e.g., MIT Design Tool, ChopChop, or CRISPick) to obtain on-target efficiency scores.
- Specificity Filtering: Use the same tools to cross-reference candidates against the reference genome, filtering out those with significant off-target potential (≤3 mismatches, especially in seed region).
- Functional Selection:
- For CRISPRko: Prioritize sgRNAs targeting early constitutive exons to maximize frameshift potential.
- For CRISPRi/a: Prioritize sgRNAs targeting regions -50 to +300 bp from the annotated Transcription Start Site (TSS).
- Final Selection: Select 3-5 top-ranked sgRNAs per target for experimental validation to account for prediction inaccuracies.
Title: sgRNA Design Workflow for CRISPRko/i/a
Effector Domains: Defining the Functional Output
The effector domain fused to a programmable DNA-binding protein (e.g., dCas9) dictates the epigenetic or catalytic outcome on the target locus.
Effector Domains by Modality
Table 2: Key Effector Domains for CRISPRko, i, and a
| Modality | Core Effector | Key Domain(s) | Molecular Function | Primary Outcome |
|---|---|---|---|---|
| CRISPRko | Wild-type Cas9 | RuvC, HNH (nuclease) | Creates DNA double-strand breaks (DSBs) | NHEJ/HDR-mediated indels, gene knockout. |
| CRISPRi | dCas9 (nuclease dead) | KRAB, SID4X (repressor) | Recruits heterochromatin factors, inhibits Pol II. | Transcriptional repression, gene knockdown. |
| CRISPRa | dCas9 (nuclease dead) | VPR, SunTag-p65-HSF1 (activator) | Recruits transcriptional co-activators (p65, Rta, VP64). | Transcriptional upregulation, gene activation. |
Protocol: Cloning and Validating Effector Constructs
Objective: Clone a dCas9-effector fusion plasmid and validate its function.
- Vector Preparation: Linearize a dCas9 backbone plasmid (e.g., pAC154-dual-dCas9-VPR) using appropriate restriction enzymes or perform Gibson Assembly/ Golden Gate cloning.
- Effector Insertion: Amplify the effector domain (e.g., KRAB for i, VPR for a) via PCR with overhangs homologous to the linearized vector. For CRISPRko, use a wild-type Cas9 plasmid (e.g., lentiCRISPRv2).
- Assembly & Transformation: Perform Gibson Assembly. Transform the reaction into competent E. coli (e.g., Stbl3). Plate on selective antibiotic agar.
- Colony Screening: Pick colonies, miniprep DNA, and validate by restriction digest and Sanger sequencing.
- Functional Validation (Reporter Assay): a. Seed HEK293T cells in a 24-well plate. b. Co-transfect with: (i) the dCas9-effector plasmid, and (ii) a reporter plasmid containing a GFP gene under a minimal promoter with an upstream sgRNA target site. c. Include controls: dCas9-only (no effector) and non-targeting sgRNA. d. After 48-72 hours, analyze GFP mean fluorescence intensity via flow cytometry. e. Expected Result: CRISPRi (KRAB) should reduce GFP signal vs. dCas9-only; CRISPRa (VPR) should increase it.
Title: Effector Domains Drive CRISPRko, i, a Outcomes
Delivery Systems: Enabling Cellular Perturbation
Efficient delivery is critical for introducing CRISPR components into target cells.
Delivery Modalities Comparison
Table 3: Comparison of Key Delivery Systems for CRISPR Components
| System | Typical Cargo | Max Capacity | Primary Cell Types | Key Advantage | Key Limitation |
|---|---|---|---|---|---|
| Lentivirus (LV) | sgRNA + Effector (plasmid or packaged) | ~8 kb | Dividing & non-dividing (e.g., neurons, macrophages) | Stable genomic integration, high efficiency. | Insertional mutagenesis risk, long-term expression. |
| Adeno-Associated Virus (AAV) | sgRNA or SaCas9 (smaller Cas9s) | ~4.7 kb | In vivo delivery (e.g., liver, eye, CNS) | Low immunogenicity, good tissue tropism. | Small cargo limit, potential pre-existing immunity. |
| Lipid Nanoparticles (LNP) | sgRNA/Cas9 mRNA or RNP | >100 nm size limit | Primary cells, in vivo (e.g., hepatocytes) | Transient expression, high efficiency in vivo, low immunogenicity. | Cell-type specific optimization needed, potential cytotoxicity. |
| Electroporation | RNP (pre-complexed sgRNA + Cas9 protein) | N/A | Immune cells (T cells, NK cells), stem cells | Rapid action, minimal off-targets, no vector DNA. | Requires specialized equipment, cell viability impact. |
Protocol: Lentiviral Production for Stable Cell Line Generation
Objective: Produce lentivirus for delivery of dCas9-effector and sgRNA.
- Day 1 - Seeding: Seed HEK293T cells (high transfection efficiency) in a 10cm dish to reach 70-80% confluence the next day.
- Day 2 - Transfection (Calcium Phosphate or PEI): a. Prepare DNA mix in sterile tube: Transfer Plasmid (e.g., pLV-dCas9-KRAB-Puro): 10 µg; Packaging Plasmids (psPAX2): 7.5 µg; (pMD2.G): 2.5 µg. b. Add CaCl₂ solution to DNA mix. Dropwise add 2X HEPES-buffered saline (HBS) while vortexing. Incubate 15 min at RT. c. Add precipitate dropwise to cells with fresh medium. Swirl gently.
- Day 3 - Medium Change: Replace medium with fresh, complete growth medium.
- Day 4 & 5 - Harvest: Collect virus-containing supernatant at 48h and 72h post-transfection. Filter through a 0.45 µm PES filter. Aliquot and store at -80°C or concentrate via ultracentrifugation.
- Day 6 - Transduction: Incubate target cells (e.g., K562) with viral supernatant plus polybrene (8 µg/mL). Spinfect at 800 x g for 30 min at 32°C. After 48h, begin selection with appropriate antibiotic (e.g., Puromycin).
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents and Materials for CRISPRko/i/a Experiments
| Item | Supplier Examples | Function in Experiment |
|---|---|---|
| dCas9-Effector Plasmids | Addgene (#107434 for dCas9-KRAB, #114195 for dCas9-VPR) | Provides the programmable DNA-binding protein fused to repressor/activator domains. |
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Addgene (#12260, #12259) | Supplies viral structural and envelope proteins for lentivirus production. |
| Lipofectamine 3000 or PEI Max | Thermo Fisher, Polysciences | Chemical transfection reagent for plasmid delivery into packaging or target cells. |
| Polybrene | Sigma-Aldrich | Cationic polymer that enhances viral transduction efficiency by neutralizing charge repulsion. |
| Puromycin Dihydrochloride | Gibco, Sigma-Aldrich | Antibiotic for selecting cells successfully transduced with plasmids containing the puromycin resistance gene. |
| Nucleofector Kit for Primary Cells | Lonza | Electroporation system and optimized buffers for delivering RNP or mRNA into hard-to-transfect cells. |
| Alt-R S.p. Cas9 Nuclease V3 | IDT | High-purity, recombinant Cas9 protein for forming RNP complexes for CRISPRko with minimal off-targets. |
| T7 Endonuclease I or NEXTGEN Indel Detection Kit | NEB, IDT | Enzyme/Kits for detecting CRISPR-induced indel mutations via mismatch cleavage or sequencing. |
| qPCR Assays for Gene Expression | Thermo Fisher, Bio-Rad | Primers/probes for quantifying mRNA levels to validate CRISPRi (knockdown) or CRISPRa (activation) efficacy. |
Experimental Design Guide: Setting Up Your CRISPR Perturbation Assay
The choice between CRISPR knockout (CRISPRko), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa) is not merely technical but strategic. This decision sits at the heart of a broader thesis on functional genomics: each modality interrogates gene function through a distinct lens, yielding complementary yet fundamentally different biological insights. CRISPRko, mediated by Cas9-induced double-strand breaks, provides a permanent, complete loss-of-function. In contrast, CRISPRi (using catalytically dead Cas9 fused to a repressive KRAB domain) and CRISPRa (using dCas9 fused to transcriptional activators like VPR) offer reversible, tunable, and often more physiologically relevant modulation of gene expression. The selection of the appropriate tool is governed by project-specific goals—ranging from identifying essential genes for oncology targets to mapping subtle regulatory networks in neurobiology—and the specific phenotype under investigation, be it cell viability, differentiation, or drug response.
Quantitative Comparison of Core Modalities
The following table summarizes the key operational and performance characteristics of CRISPRko, CRISPRi, and CRISPRa based on current literature and benchmark studies.
Table 1: Core Characteristics of CRISPRko, CRISPRi, and CRISPRa
| Feature | CRISPRko (e.g., Cas9) | CRISPRi (e.g., dCas9-KRAB) | CRISPRa (e.g., dCas9-VPR) |
|---|---|---|---|
| Catalytic Activity | Active endonuclease (creates DSBs) | Catalytically dead; repressive fusion | Catalytically dead; activating fusion |
| Primary Effect | Indels → Frameshifts/Nonsense mutations → Protein ablation | Epigenetic repression → Reduced transcription | Epigenetic activation → Increased transcription |
| Reversibility | Permanent | Reversible (upon sgRNA/dCas9 removal) | Reversible (upon sgRNA/dCas9 removal) |
| Knockdown Efficiency | Typically >80% protein loss | Typically 70-95% mRNA reduction | Typically 2-10x mRNA induction (varies widely) |
| Kinetics | Fast (protein loss depends on turnover) | Fast (repression within hours) | Fast (activation within hours) |
| Off-Target Effects | DNA cleavage at off-target sites | Minimal; transcriptional repression at off-target sites | Minimal; transcriptional activation at off-target sites |
| Key Advantage | Complete loss-of-function; gold standard for essentiality screens | Tunable, reversible; avoids confounding DNA damage response | Gain-of-function; studies gene overexpression phenotypes |
| Key Limitation | Confounds from DNA damage response/p53 activation; clone outgrowth | Repression may be incomplete; position-dependent efficiency | Activation is highly context- and locus-dependent |
| Ideal Use Case | Identifying essential genes; studying null phenotypes; targeting non-coding regions | Hypomorphic studies; essential gene network mapping; sensitive cell types | Screening for gene overexpression effects; rescuing knockdowns; studying enhancers |
Strategic Selection: Aligning Goals with Modality
Project Goal-Driven Selection
- Identification of Essential Genes & Drug Targets (Fitness Screens): CRISPRko is the predominant, robust choice for genome-wide loss-of-function screens to identify genes required for cell survival/proliferation. Its permanent effect is ideal for long-term assays. CRISPRi is a powerful alternative, especially in cells sensitive to DNA damage or where a titratable knockdown is desired to avoid synthetic lethality from complete knockout.
- Mapping Gene Regulatory Networks & Synthetic Lethality: CRISPRi excels here. Its reversibility and tunability allow for the study of gene interactions, partial knockdown phenotypes, and essential gene networks without triggering apoptosis. Paired CRISPRi/a can be used to probe dose-dependent relationships.
- Gain-of-Function Screening & Cellular Programming: CRISPRa is the exclusive tool for systematic overexpression screens, useful for identifying genes that confer drug resistance, drive differentiation, or overcome tumor suppressor effects.
Phenotype-Driven Selection
- Viability/Proliferation: All three are applicable, but CRISPRko is standard. Use CRISPRi for more nuanced, time-sensitive viability phenotypes.
- Differentiation & Development: CRISPRa and CRISPRi are superior due to their reversibility and ability to model subtle, temporal changes in gene expression without genotoxic stress.
- Drug Response (Resistance/Sensitivity): CRISPRko for identifying loss-of-resistance mechanisms; CRISPRa for identifying overexpression-driven resistance.
- Sensitive Cell Types (e.g., Primary, Neurons): CRISPRi/a is often preferred to avoid the cytotoxic stress and clonal heterogeneity introduced by double-strand breaks from CRISPRko.
Detailed Methodologies for Key Experiments
Protocol: Genome-wide CRISPRko Fitness Screen
Objective: Identify genes essential for cell proliferation/survival.
- Library Transduction: Transduce target cells (at MOI ~0.3) with a lentiviral pooled sgRNA library (e.g., Brunello, ~4 sgRNAs/gene, 75k sgRNAs total). Include a non-targeting control sgRNA set.
- Selection & Expansion: Treat cells with puromycin (1-2 µg/mL) for 5-7 days to select for transduced cells. Harvest an initial reference timepoint (T0). Propagate remaining cells for 14-21 population doublings (Tfinal).
- Genomic DNA Extraction & PCR Amplification: Isolate gDNA from T0 and Tfinal pellets (≥50µg). Amplify integrated sgRNA sequences via two-step PCR using indexing primers to add sequencing adapters and sample barcodes.
- Next-Generation Sequencing & Analysis: Sequence PCR products on an Illumina platform. Align reads to the reference sgRNA library. Use MAGeCK or similar algorithms to compare sgRNA abundance between T0 and Tfinal, identifying significantly depleted sgRNAs/genes.
Protocol: CRISPRi/a Transcriptional Modulation Validation
Objective: Validate target gene knockdown (CRISPRi) or upregulation (CRISPRa) prior to a functional screen.
- sgRNA Design & Cloning: Design 3-5 sgRNAs targeting the promoter region near the TSS (for CRISPRi, -50 to +300 bp relative to TSS; for CRISPRa, -400 to -50 bp). Clone into appropriate dCas9-effector vector.
- Cell Line Engineering: Stably express dCas9-KRAB (CRISPRi) or dCas9-VPR (CRISPRa) in target cells via lentiviral transduction and blasticidin selection.
- Transfection/Transduction of sgRNAs: Deliver sgRNA vectors into the engineered cell line. Include non-targeting and targeting controls (e.g., for a known essential gene in CRISPRi).
- qRT-PCR Analysis: 72-96 hours post-sgRNA delivery, extract RNA, synthesize cDNA, and perform qPCR for the target gene. Normalize to housekeeping genes (e.g., GAPDH, ACTB). Calculate fold-change relative to non-targeting sgRNA control.
Visualizing Selection Logic and Workflows
Diagram 1: CRISPR Modality Selection Logic Flow
Diagram 2: Generic Pooled CRISPR Screen Protocol
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for CRISPR Functional Genomics
| Reagent / Material | Function & Rationale | Example/Catalog Consideration |
|---|---|---|
| Validated sgRNA Library | Pre-designed, pooled sets of sgRNAs ensuring genome-wide coverage and high on-target efficiency. Essential for reproducible screens. | Brunello (CRISPRko), Dolcetto (CRISPRi), Calabrese (CRISPRa) from Addgene. |
| Lentiviral Packaging Plasmids | For safe, efficient production of lentiviral particles carrying CRISPR machinery (Cas9/dCas9) and sgRNAs. | psPAX2 (packaging) and pMD2.G (VSV-G envelope) are standard. |
| dCas9-Effector Plasmid | Expresses the catalytically dead Cas9 fused to transcriptional modulators. The core of CRISPRi/a. | pHAGE dCas9-KRAB (CRISPRi) or pHAGE dCas9-VPR (CRISPRa). |
| Stable Cell Line Reagents | Antibiotics for selecting and maintaining cells expressing Cas9/dCas9. Critical for screen consistency. | Puromycin, Blasticidin, or Hygromycin B, depending on resistance markers. |
| Next-Generation Sequencing Kit | For preparing sgRNA amplicon libraries from genomic DNA for deep sequencing. | Illumina-compatible kits (e.g., NEBNext). Indexing primers are critical. |
| Genomic DNA Extraction Kit | High-yield, high-purity gDNA extraction is vital for accurate representation of sgRNA abundance. | Kits optimized for cultured mammalian cells (e.g., Qiagen Blood & Cell Culture DNA Kit). |
| Analysis Software Pipeline | Computational tool to quantify sgRNA read counts and perform statistical analysis of enrichment/depletion. | MAGeCK, PinAPL-Py, or CRISPRcloud. |
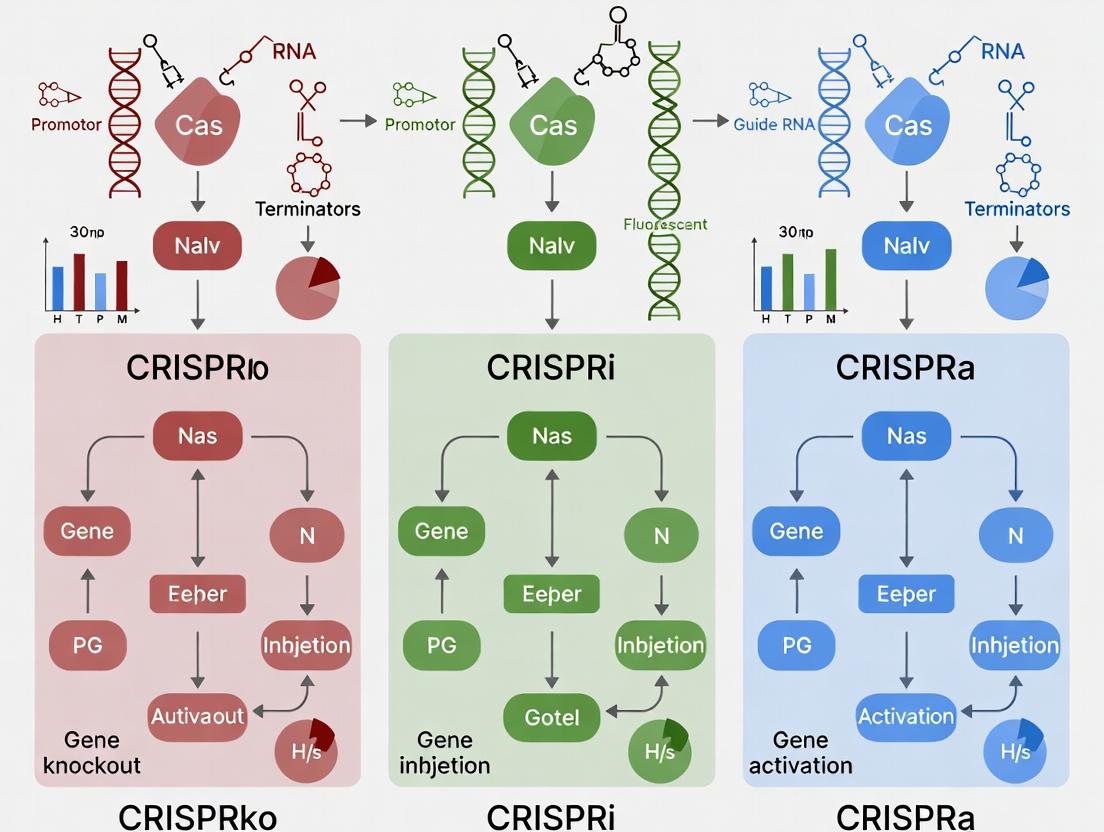
This technical guide, framed within a broader thesis comparing CRISPR knock-out (CRISPRko), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa), provides a detailed, step-by-step protocol from initial construct design to final transfection. These technologies, while sharing a common Cas protein origin, diverge significantly in their mechanisms—permanent gene disruption, transcriptional repression, and transcriptional activation, respectively—leading to distinct experimental workflows and reagent requirements. This guide is designed for researchers and drug development professionals implementing these precise genome-modulation tools.
The fundamental difference between CRISPRko, i, and a lies in the nuclease activity of the Cas protein and the fusion of effector domains.
- CRISPRko: Utilizes wild-type Cas9 (or Cas12a) to create double-strand breaks, leading to frameshift mutations and gene knock-out via non-homologous end joining (NHEJ).
- CRISPRi: Employs a catalytically dead Cas9 (dCas9) fused to a transcriptional repressor domain (e.g., KRAB). It binds to the target DNA without cutting, blocking transcription initiation or elongation.
- CRISPRa: Uses dCas9 fused to a transcriptional activator domain (e.g., VP64, p65AD). It recruits the cellular transcription machinery to the promoter region to upregulate gene expression.
Key Research Reagent Solutions
| Reagent Category | Specific Item (Example) | Function in CRISPRko/i/a |
|---|---|---|
| Core Nuclease/Effector | SpCas9 Nuclease (WT) | CRISPRko: Creates DSBs for gene disruption. |
| dCas9-KRAB Plasmid | CRISPRi: DNA-binding platform for transcriptional repression. | |
| dCas9-VP64 Plasmid | CRISPRa: DNA-binding platform for transcriptional activation. | |
| Guide RNA (gRNA) | Synthetic sgRNA (chemically modified) | Directs Cas/dCas protein to the specific genomic target sequence. |
| gRNA Expression Cloning Kit | For cloning gRNA sequences into U6 or other Pol III promoter vectors. | |
| Delivery System | Lipofectamine CRISPRMAX | Lipid nanoparticles for efficient ribonucleoprotein (RNP) or plasmid delivery. |
| Lentiviral Packaging Mix (psPAX2, pMD2.G) | For creating stable cell lines via viral transduction (common for CRISPRi/a). | |
| Validation & Selection | SURVEYOR or T7E1 Assay Kit | Detects indels formed by NHEJ after CRISPRko. |
| Puromycin Dihydrochloride | Selection antibiotic for cells successfully transduced with lentiviral constructs. | |
| qPCR Assay for Target Gene | Quantifies changes in mRNA expression levels for CRISPRi and CRISPRa. |
Step-by-Step Protocol Comparison
The following tables outline the critical differences in protocol from design to analysis.
Table 1: Construct Design & Cloning
| Step | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| 1. Target Selection | Exons early in the coding sequence to maximize frameshift probability. | Promoter or 5' UTR regions, typically -50 to +300 bp relative to TSS. | Promoter regions upstream of TSS, often -400 to -50 bp. |
| 2. gRNA Design | Prioritize on-target efficiency (predictive algorithms) and minimize off-targets. | Also consider chromatin accessibility and avoid transcription factor binding sites. | Similar to CRISPRi; some systems (e.g., SAM) use 2-3 gRNAs for synergy. |
| 3. Effector Vector | Wild-type Cas9 (SpCas9, SaCas9) expression plasmid or mRNA. | dCas9-KRAB fusion expression construct. | dCas9-activator fusion (e.g., dCas9-VP64-p65-Rta (VPR)). |
| 4. Cloning Strategy | Clone sgRNA into a U6-driven vector; can be separate from or combined with Cas9. | Clone sgRNA into a Pol III promoter vector, often part of a lentiviral all-in-one system with dCas9-KRAB and a puromycin marker. | Clone single or multiple sgRNAs into vectors compatible with the chosen activation system (e.g., SAM requires MS2 stem-loops in gRNA). |
Table 2: Delivery & Transfection
| Step | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Primary Method | Transient: RNP (Cas9 protein + sgRNA) or plasmid co-transfection. High efficiency, quick turnover. | Stable: Lentiviral transduction of dCas9-KRAB cell line, followed by lentiviral sgRNA delivery. Ensures uniform, persistent repression. | Stable: Similar to CRISPRi. Often requires generation of a stable dCas9-activator cell line first. |
| Typical Format | Plasmid(s): 2 µg Cas9 + 1 µg gRNA plasmid per well (24-well). RNP: 20 pmol Cas9 + 40 pmol sgRNA. | Lentivirus: Transduce at MOI ~3-10 to create polyclonal dCas9-KRAB line. Select with puromycin (1-5 µg/mL) for 5-7 days. | Lentivirus: Similar MOI. Selection conditions depend on the specific activator construct's resistance markers. |
| Critical Control | Non-targeting sgRNA control. Transfection reagent-only control. | Non-targeting sgRNA control. Wild-type (no dCas9) cell control. | Non-targeting sgRNA control. Optional: Known positive activation target (e.g., MYOD1). |
Table 3: Post-Transfection Analysis & Validation
| Step | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Timeline | Analyze 48-72 hours post-transfection (RNP) or 3-5 days (plasmid). | Assay ≥5 days post-sgRNA transduction to allow for protein turnover and epigenetic effects. | Assay ≥5 days post-sgRNA transduction; maximal activation may take up to 2 weeks. |
| Primary Validation | Indel Detection: T7E1 assay, Sanger sequencing with decomposition tools (TIDE, ICE), or NGS. | mRNA Downregulation: RT-qPCR (most direct). Protein analysis via Western blot or flow cytometry. | mRNA Upregulation: RT-qPCR. Protein analysis. |
| Secondary Analysis | Phenotypic assays (proliferation, survival). Confirm loss of protein via Western/Flow. | RNA-seq for genome-wide expression changes and off-target effects. ChIP-seq for dCas9-KRAB binding. | RNA-seq to assess specificity and magnitude of activation. |
| Key Metric | Indel % (typically >70% for efficient KO). | % Repression (often 60-95% for robust targets). | Fold Activation (highly variable; 2-100x depending on target and system). |
Detailed Experimental Protocol: Lentiviral CRISPRi/a Stable Cell Line Generation
This is a critical shared workflow for CRISPRi and CRISPRa applications requiring sustained modulation.
Protocol:
- Day 1: Seed HEK293T packaging cells in a 6-well plate (70% confluency) in DMEM + 10% FBS, no antibiotics.
- Day 2: Co-transfect cells using a polyethylenimine (PEI) protocol:
- Prepare DNA mix in 150 µL Opti-MEM: 1.5 µg of lentiviral effector plasmid (dCas9-KRAB or dCas9-VPR), 1.0 µg of psPAX2 (packaging plasmid), and 0.5 µg of pMD2.G (envelope plasmid).
- Prepare PEI mix: 9 µL of 1 mg/mL PEI in 150 µL Opti-MEM.
- Combine DNA and PEI mixes, incubate 15 min at RT, add dropwise to cells.
- Day 3: Replace media with 2 mL fresh complete growth media.
- Day 4 & 5: Harvest viral supernatant (~48 and 72 hours post-transfection), filter through a 0.45 µm PES filter, and either use immediately or aliquot and store at -80°C.
- Day 5: Transduction of Target Cells. Seed your target cell line (e.g., K562, HeLa) in a 24-well plate. Thaw viral supernatant and add to cells with polybrene (final concentration 8 µg/mL). Spinoculate by centrifuging plates at 800 x g for 30-60 min at 32°C, then incubate.
- Day 6: Replace media with fresh growth media.
- Day 7-14: Begin antibiotic selection (e.g., 2 µg/mL puromycin for dCas9-KRAB). Maintain selection for 5-7 days until all cells in an untransduced control well have died.
- Validation: Validate dCas9 expression in the polyclonal population via Western blot or functional test (transduce with a control sgRNA and assay by qPCR).
Visual Workflows
Comparison of CRISPRko, i, and a experimental workflows.
Molecular mechanisms differentiating CRISPRko, CRISPRi, and CRISPRa.
The selection of CRISPR knockout (CRISPRko), CRISPR interference (CRISPRi), or CRISPR activation (CRISPRa) technologies fundamentally shapes the design, interpretation, and application of functional genomic screens. CRISPRko provides complete loss-of-function, enabling the identification of essential genes under positive selection. CRISPRi offers tunable, reversible knockdown, ideal for studying dosage-sensitive genes and essential gene networks. CRISPRa allows for gain-of-function and gene overexpression, facilitating the discovery of tumor suppressors and genes conferring phenotypic resistance. This guide details the technical application of each modality within large-scale screening frameworks for target discovery.
Quantitative Comparison of Core Technologies
The following table summarizes the key quantitative and functional characteristics of each system for screening applications.
Table 1: Comparative Analysis of CRISPRko, CRISPRi, and CRISPRa for Genomic Screens
| Parameter | CRISPRko (Knockout) | CRISPRi (Interference) | CRISPRa (Activation) |
|---|---|---|---|
| Core Mechanism | NHEJ-mediated indels causing frameshifts and premature stop codons. | dCas9 fused to transcriptional repressor domains (e.g., KRAB). | dCas9 fused to transcriptional activator domains (e.g., VPR, SAM). |
| Effect on Gene | Permanent, complete loss-of-function. | Reversible, tunable transcriptional repression (typically 70-95% knockdown). | Transcriptional overexpression (often 2-10x+ induction). |
| Typical Screening Library | Whole-genome (e.g., Brunello, Brie), sub-library (e.g., kinome). | CRISPRi-v2 (hg38) with optimized sgRNAs for TSS repression. | CRISPRa-v2 (hg38) with sgRNAs designed for promoter-proximal targeting. |
| Optimal Targeting Region | Early exons of the coding sequence. | -50 to +300 bp relative to the Transcription Start Site (TSS). | -200 to +50 bp relative to the TSS. |
| Key Application in Screens | Identification of essential genes (positive selection), synthetic lethality. | Hypomorphic studies, essential gene network analysis, long-term phenotypic assays. | Identification of genes whose overexpression confers resistance or a phenotype (negative selection). |
| Primary Readout | Depletion of sgRNAs in a viability screen. | Depletion (for essential genes) or enrichment (for suppressor screens) of sgRNAs. | Enrichment of sgRNAs conferring a survival or resistance advantage. |
| Data Analysis Tool | MAGeCK, BAGEL, CERES (to correct for copy-number effects). | MAGeCK, PinAPL-Py. | MAGeCK, drugZ. |
Detailed Experimental Protocols for Screening
Protocol A: Pooled Lentiviral CRISPRko Screen for Essential Genes
Objective: Identify genes essential for cell proliferation/survival.
- Library Selection & Cloning: Select a genome-scale sgRNA library (e.g., Brunello, ~4 sgRNAs/gene). Amplify the plasmid library and prepare high-quality lentiviral packaging mix (psPAX2, pMD2.G).
- Viral Production & Titering: Produce lentivirus in HEK293T cells. Determine viral titer via puromycin kill curve or GFP expression to achieve an MOI ~0.3-0.4, ensuring >500x library representation.
- Cell Infection & Selection: Infect target cells (e.g., a cancer cell line) at scale. Apply puromycin (1-5 µg/mL) 24h post-infection for 5-7 days to select transduced cells.
- Screen Harvest: At selection end (Day 0), harvest a baseline population (≥50M cells, 500x coverage). Split remaining cells into replicate pools and culture for ~14 population doublings. Harvest final population (Day 14).
- Genomic DNA Extraction & Sequencing: Extract gDNA (Qiagen Maxi Prep). Perform a two-step PCR: (i) Amplify integrated sgRNA cassettes from gDNA (20-30 cycles); (ii) Add Illumina adaptors and sample barcodes (10-12 cycles). Pool and sequence on an Illumina HiSeq/NovaSeq (≥100 reads/sgRNA).
- Data Analysis: Align sequences to the reference library. Count reads per sgRNA for Day 0 and Day 14 samples. Use MAGeCK (v0.5.9) to test for significant sgRNA depletion and rank essential genes (FDR < 0.05).
Protocol B: CRISPRi/a Screening for Modulating Drug Response
Objective: Identify genes whose repression (CRISPRi) or activation (CRISPRa) alter sensitivity to a therapeutic compound.
- Stable Cell Line Generation: Create a cell line stably expressing dCas9-KRAB (for CRISPRi) or dCas9-VPR (for CRISPRa) via lentiviral transduction and blasticidin selection.
- Specialized Library Transduction: Transduce the stable line with the appropriate CRISPRi-v2 or CRISPRa-v2 library, following steps in Protocol A for infection and puromycin selection.
- Compound Challenge: Post-selection, split cells into two arms: DMSO vehicle control and Drug-treated (at IC50-IC70 concentration). Maintain cells for 10-14 doublings, replenishing compound/DMSO every 2-3 days. Maintain 500x library coverage throughout.
- Harvest & Sequencing: Harvest cells from all conditions at endpoint. Process gDNA and prepare sequencing libraries as in Protocol A.
- Analysis of Modulators: Use MAGeCK or drugZ to compare sgRNA abundances between drug-treated and control arms. For CRISPRi: sgRNAs enriched in drug treatment indicate sensitizers (gene knockdown increases drug efficacy); depleted sgRNAs indicate resistors. For CRISPRa: Enriched sgRNAs indicate suppressors (overexpression confers resistance).
Diagrams of Screening Workflows and Pathways
Diagram 1: CRISPRko/i/a Screening Workflow
Diagram 2: Core Mechanisms of CRISPRko, i, and a
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Reagents for CRISPR Functional Genomic Screens
| Reagent/Material | Function & Critical Notes |
|---|---|
| Validated sgRNA Library | Pre-designed, pooled plasmid libraries (e.g., Brunello for ko, CRISPRi-v2, CRISPRa-v2). Ensures specificity and coverage. |
| Lentiviral Packaging Plasmids | psPAX2 (gag/pol) and pMD2.G (VSV-G envelope). Required for production of replication-incompetent lentivirus. |
| HEK293T Cells | Standard cell line for high-titer lentivirus production due to high transfection efficiency. |
| Polybrene (Hexadimethrine Bromide) | Cationic polymer used during transduction to enhance viral attachment and entry (typical use: 4-8 µg/mL). |
| Selection Antibiotics | Puromycin (for sgRNA vector selection), Blasticidin (for dCas9 stable line selection). Concentration must be pre-titrated. |
| High-Quality gDNA Extraction Kit | Scalable kit for large cell pellets (e.g., Qiagen Blood & Cell Culture Maxi Kit). High yield/purity is critical for PCR. |
| Q5 High-Fidelity DNA Polymerase | Used for sgRNA amplicon PCR to minimize amplification errors and bias during NGS library prep. |
| Dual-Indexed Illumina Primers | Custom primers for the second-step PCR to multiplex multiple screening conditions on one sequencing run. |
| Cell Counter & Size Analyzer | Automated cell counter (e.g., Beckman Coulter Vi-CELL). Essential for accurately determining cell numbers to maintain library representation. |
| Cas9/dCas9-Expressing Cell Line | For CRISPRi/a, a clonal line with stable, uniform expression of the dCas9 fusion protein is foundational. |
Introduction Within the framework of a systematic thesis comparing CRISPRko (knockout), CRISPRi (interference), and CRISPRa (activation), the selection of an appropriate biological model is paramount. The functional output and interpretation of these orthogonal CRISPR modalities are profoundly influenced by the cellular context, transcriptional state, and system complexity. This technical guide details the application of advanced models—induced pluripotent stem cells (iPSCs), organoids, and in vivo systems—in CRISPR perturbation screens, highlighting model-specific protocols, data considerations, and reagent toolkits.
iPSC Models: Defining Fundamental Genetic Networks
Human iPSCs provide a genetically defined, renewable platform for studying gene function in development and disease. Their compatibility with precise genome editing makes them ideal for head-to-head comparisons of CRISPRko/i/a. CRISPRi and CRISPRa are particularly powerful here for probing dosage-sensitive genes and developmental pathways where complete knockout may be lethal or impede differentiation.
Experimental Protocol: CRISPRi/a Differentiation Screen in iPSCs
- Engineered iPSC Line Generation: Stably express dCas9-KRAB (for CRISPRi) or dCas9-VPR (for CRISPRa) in a safe-harbor locus (e.g., AAVS1) using a PiggyBac transposon or homology-directed repair.
- Library Delivery: Transduce the engineered line at low MOI (<0.3) with a lentiviral sgRNA library targeting a gene set of interest (e.g., transcription factors). Maintain >500x coverage.
- Selection & Differentiation: Puromycin-select for sgRNA+ cells for 5-7 days. Split cells and initiate a directed differentiation protocol (e.g., to cortical neurons, cardiomyocytes) for 2-4 weeks.
- Phenotypic Sorting: Harvest cells at relevant time points. Use FACS to isolate populations based on differentiation markers (e.g., SOX2+ progenitors, TUJ1+ neurons).
- Genomic DNA Extraction & NGS: Extract gDNA from sorted populations and the plasmid library pool. Perform a two-step PCR to add sequencing adapters and barcodes. Sequence on an Illumina platform.
- Analysis: Align reads to the sgRNA library. Use MAGeCK or similar tools to compare sgRNA abundance between conditions (e.g., differentiated vs. undifferentiated) to identify hits that promote or block differentiation.
Table 1: Quantitative Performance of CRISPR Modalities in iPSC Neurogenesis Screen
| Modality | Target Genes Screened | Hit Rate (FDR < 0.1) | Avg. Log2 Fold Change (Top Hit) | Key Advantage in iPSCs |
|---|---|---|---|---|
| CRISPRko | 200 (Essential Genes) | 12% | -4.2 | Unambiguous loss-of-function; identifies absolute必需品 |
| CRISPRi | 200 (Polycomb Targets) | 18% | -2.8 | Tunable, reversible suppression; minimal differentiation block |
| CRISPRa | 200 (Developmental TFs) | 9% | +3.5 | Activates silent loci; probes gain-of-function in naive state |
The Scientist's Toolkit: iPSC CRISPR Screening
| Reagent/Material | Function |
|---|---|
| dCas9-KRAB-iPSC Line | Enables stable, inducible transcriptional repression (CRISPRi). |
| Lentiviral sgRNA Library | Delivers pooled genetic perturbations; format varies (CRISPRko/i/a). |
| mTeSR Plus Medium | Feeder-free, defined medium for maintaining iPSC pluripotency. |
| Y-27632 (ROCK inhibitor) | Improves viability after dissociation (passaging or sorting). |
| Accutase | Gentle enzyme for harvesting iPSCs as single cells. |
| Differentiation Kit (e.g., Cardiomyocyte) | Provides standardized protocols and reagents for lineage commitment. |
Diagram 1: iPSC CRISPR Screen Workflow
Organoid Models: Probing Tissue-Level Phenotypes
Organoids recapitulate tissue architecture and cell-cell interactions, offering a middle ground for studying gene function in a structured microenvironment. CRISPRko is critical for modeling tumor suppressor loss, while CRISPRi/a can modulate pathways controlling morphogenesis or cell fate patterning without ablating entire cell populations.
Experimental Protocol: CRISPRko in Cerebral Organoids for Tumor Modeling
- sgRNA Electroporation: Introduce plasmids expressing Cas9 and tumor-specific sgRNAs (e.g., targeting TP53, PTEN) into iPSCs via nucleofection.
- Organoid Initiation: Aggregate edited iPSCs in low-adherence V-bottom plates to form embryoid bodies. Transfer to Matrigel droplets and culture in neural induction medium.
- Long-term Culture: Maintain organoids in spinning bioreactors or orbital shakers for 2-6 months, feeding with differentiation media.
- Phenotypic Analysis: Monitor for overgrowth or structural aberrations. Perform:
- Immunohistochemistry: Section and stain for proliferation (Ki67), neural markers (SOX2, TUJ1), and ectopic expression.
- Bulk/Single-cell RNA-seq: Profile transcriptional changes.
- Imaging: Confocal microscopy for 3D structure.
- Validation: Isolve genomic DNA from abnormal regions for sequencing to confirm edits and off-target analysis.
Table 2: Phenotypic Outcomes in Cerebral Organoid CRISPR Screen
| Perturbation (Modality) | Target Gene | Readout (Day 60) | Quantified Metric |
|---|---|---|---|
| CRISPRko | TP53 | Increased Progenitor Zone | Ki67+ area increased by 45% (±8%) |
| CRISPRko | PTEN | Enlarged Organoid Size | Diameter increased by 2.3x (±0.4x) |
| CRISPRi | CDKN2C | Altered Cell Cycle | G1 phase reduced by 22% (±5%) |
| CRISPRa | MYC | Hyperproliferation | Scattered SOX2+ clusters |
The Scientist's Toolkit: Organoid CRISPR Engineering
| Reagent/Material | Function |
|---|---|
| Nucleofector Kit for iPSCs | High-efficiency delivery of CRISPR RNP or plasmid DNA. |
| Growth Factor-Reduced Matrigel | Provides a 3D extracellular matrix for organoid development. |
| Spinning Bioreactor | Improves nutrient/waste exchange for long-term organoid culture. |
| Tissue-Tek OCT Compound | For embedding organoids for cryosectioning. |
| Confocal Imaging Dish | Glass-bottom dishes for high-resolution 3D live imaging. |
Diagram 2: Organoid CRISPR Tumor Modeling
In Vivo Systems: Validating Functional Impact
In vivo models provide the ultimate context for studying gene function, incorporating systemic physiology and immune interactions. CRISPRko screens in vivo are well-established in immunology and oncology. CRISPRi/a enables tissue-specific, inducible, and reversible perturbations to model disease progression or therapeutic intervention.
Experimental Protocol: In Vivo CRISPRko Screen in Patient-Derived Xenografts (PDX)
- Library Preparation: Generate a Cas9-expressing tumor cell line (PDX-derived or cell line). Transduce with a focused sgRNA library (e.g., kinome).
- Engraftment: Inject 1-5x10^6 library-cells subcutaneously or orthotopically into immunocompromised mice (NSG). Maintain >500x representation.
- Tumor Growth & Treatment: Monitor tumor volume. Optionally, administer a drug treatment to identify resistance/sensitivity genes.
- Harvest & Processing: Collect tumors at endpoint (e.g., volume ~1500 mm³). Split sample: one part for gDNA extraction, another for frozen/FFPE sectioning.
- Sequencing & Analysis: Isolate gDNA from tumor and the pre-injection cell pool. Prepare NGS libraries. Analyze for sgRNA enrichment/depletion to identify genes affecting tumor growth or drug response.
Table 3: In Vivo Screen Results for Chemotherapy Resistance
| sgRNA Target (Gene) | Pre-injection Abundance (%) | Final Tumor Abundance (%) | Log2 Fold Change | Interpretation |
|---|---|---|---|---|
| sg-Control | 0.100 | 0.095 | -0.07 | Neutral |
| sg-MDR1 | 0.100 | 0.010 | -3.32 | Sensitizing hit |
| sg-BCL2 | 0.100 | 0.300 | +1.58 | Resistance hit |
| sg-TP53 | 0.100 | 0.500 | +2.32 | Strong driver |
The Scientist's Toolkit: In Vivo CRISPR Screening
| Reagent/Material | Function |
|---|---|
| NSG (NOD-scid-IL2Rγnull) Mice | Immunodeficient host for engrafting human cells/tumors. |
| Lentiviral Concentrator | Produces high-titer virus for efficient library delivery. |
| In Vivo Imaging System (IVIS) | For bioluminescent tracking of tumor growth/metastasis. |
| DNeasy Blood & Tissue Kit | Robust gDNA extraction from heterogeneous tumor tissue. |
| PicoGreen dsDNA Assay | Accurately quantifies gDNA for equal NGS library input. |
Diagram 3: In Vivo PDX CRISPRko Screen Logic
Conclusion The strategic deployment of CRISPRko, CRISPRi, and CRISPRa across iPSC, organoid, and in vivo models enables a comprehensive dissection of gene function across scales of biological complexity. iPSCs offer precision for foundational networks, organoids reveal morphological consequences, and in vivo systems deliver physiological relevance. The choice of model must be driven by the specific biological question within the CRISPR modality thesis, as each system presents unique advantages and constraints for perturbation and readout.
CRISPRi/a for Studying Essential Genes and Dosage-Sensitive Phenotypes
The functional genomics toolkit has been revolutionized by CRISPR-Cas systems. The core thesis differentiating CRISPR knockout (CRISPRko), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa) hinges on their distinct mechanistic outputs and resultant biological applications. CRISPRko uses Cas9 nuclease to create double-strand breaks, leading to frameshift mutations and permanent gene knockout. In contrast, CRISPRi employs a catalytically dead Cas9 (dCas9) fused to transcriptional repressors (e.g., KRAB) to epigenetically silence gene expression without altering the DNA sequence. CRISPRa uses dCas9 fused to transcriptional activators (e.g., VP64, p65AD, SunTag) to upregulate gene expression.
This whitepaper focuses on CRISPRi and CRISPRa, which are uniquely suited for probing essential genes and dosage-sensitive phenotypes—areas where traditional CRISPRko is limited. Knocking out an essential gene is lethal, precluding the study of its function in cellular viability. Similarly, many biological processes and disease states (e.g., haploinsufficiency, oncogene overexpression) are sensitive to precise expression levels, not merely presence or absence. CRISPRi/a enables tunable, reversible modulation of gene expression, allowing researchers to titrate dosage and study consequent phenotypes in a controlled manner.
Core Mechanisms and Quantitative Comparisons
Mechanism of Action
CRISPRi: The dCas9-KRAB fusion is guided to a target site near the transcription start site (TSS) of a gene. The KRAB domain recruits endogenous repressive complexes (e.g., SETDB1, HP1), leading to heterochromatin formation via H3K9 trimethylation, effectively silencing transcription.
CRISPRa: Multiple architectures exist. The common dCas9-VP64 activator can be enhanced by using synergistic activation mediator (SAM) systems. The SAM system involves dCas9-VP64, a modified sgRNA with MS2 RNA aptamers, and the MS2-p65-HSF1 fusion protein, which recruits additional activators to robustly upregulate transcription.
Quantitative Performance Data
Table 1: Performance Metrics of CRISPRi vs. CRISPRa Systems
| Parameter | CRISPRi (dCas9-KRAB) | CRISPRa (dCas9-VP64) | CRISPRa (SAM System) |
|---|---|---|---|
| Typical Repression/Activation Fold-Change | 10- to 100-fold knockdown | 2- to 10-fold activation | 10- to 1,000-fold activation |
| Optimal Targeting Region | -50 to +300 bp relative to TSS | -400 to -50 bp upstream of TSS | -400 to -50 bp upstream of TSS |
| On-Target Efficiency Range | 70-95% knockdown for optimal sgRNAs | 30-70% activation (VP64 alone) | 80-95% activation for optimal sgRNAs |
| Multiplexing Capacity | High (multiple sgRNAs) | Moderate | High (with MS2-modified sgRNAs) |
| Typical Off-Target Effects | Low (transcriptional, no DNA damage) | Low | Moderate (increased transcriptional noise) |
| Reversibility | High | High | High |
Table 2: Application in Essential Gene & Dosage Studies
| Study Type | CRISPRko Suitability | CRISPRi Suitability | CRISPRa Suitability |
|---|---|---|---|
| Essential Gene Function | Low (lethal) | High (titratable knockdown, hypomorphs) | Low (overexpression may not rescue) |
| Haploinsufficiency Modeling | Low (binary) | High (mimics reduced dose) | Low (increases dose) |
| Oncogene Overexpression Studies | Low (cannot overexpress) | Low (represses) | High (models gain-of-function) |
| Tunable Dosage Response | None | High (via sgRNA/dCas9 titration) | High (via sgRNA/dCas9 titration) |
| Longitudinal/Reversible Studies | Low (permanent) | High (inducible systems) | High (inducible systems) |
Detailed Experimental Protocols
Protocol: CRISPRi Pooled Screening for Essential Genes
Objective: Identify and validate essential genes in a cancer cell line using a genome-wide CRISPRi knockdown screen.
Materials: See "Scientist's Toolkit" below.
Methodology:
- Library Design: Obtain a genome-wide CRISPRi sgRNA library (e.g., Brunello-i). sgRNAs (typically 3-5 per gene) are designed to target the -50 to +300 bp region relative to the TSS.
- Lentiviral Production: Generate lentivirus for the sgRNA library at a low MOI (<0.3) to ensure single integration in HEK293T cells using standard packaging plasmids (psPAX2, pMD2.G).
- Cell Infection and Selection: Infect target cells (e.g., A549) with the lentiviral library. Maintain a minimum of 500x library representation at all steps. Select transduced cells with puromycin (2 µg/mL) for 7 days.
- Screen Passage: Passage cells for 14-21 population doublings. Harvest genomic DNA from a minimum of 50 million cells at the initial (T0) and final (Tend) time points using a large-scale gDNA kit.
- sgRNA Amplification & Sequencing: Amplify integrated sgRNA sequences from gDNA via a two-step PCR. First PCR uses primers flanking the sgRNA scaffold. Second PCR adds Illumina adapters and sample barcodes. Pool and sequence on an Illumina NextSeq (75bp single-end).
- Data Analysis: Map reads to the sgRNA library. Use a tool like MAGeCK or CRISPResso2 to calculate sgRNA depletion scores. Essential genes are identified by significant depletion of targeting sgRNAs in the End sample compared to T0 (FDR < 0.05).
Protocol: CRISPRa for Dosage-Sensitive Phenotype Analysis
Objective: Titrate the expression of a haploinsufficient tumor suppressor gene (e.g., PTEN) and measure dose-dependent phenotypic outputs.
Materials: See "Scientist's Toolkit" below.
Methodology:
- Stable Cell Line Generation: Lentivirally transduce target cells with a stable, inducible dCas9-SAM activator system (e.g., pLV-dCas9-SAM). Select with blasticidin (10 µg/mL) for 10 days. Generate a monoclonal cell line via FACS or limiting dilution.
- sgRNA Cloning & Validation: Clone 3-5 sgRNAs targeting the -400 to -50 bp region upstream of the PTEN TSS into a lentiviral sgRNA(MS2) vector (e.g., pLX-sgRNA(MS2)-zeo). Produce individual lentiviruses and transduce the dCas9-SAM cell line. Select with zeocin (200 µg/mL) for 7 days.
- Dose Titration: To titrate expression, use two methods: A) Doxycycline Titration: Treat cells with a range of doxycycline (0, 0.1, 0.5, 1.0, 2.0 µg/mL) to induce dCas9-SAM expression differentially. B) Mixed Population: Create a pooled population with all PTEN-targeting sgRNAs; expression variance across cells provides a dose gradient.
- Phenotypic Readouts: At 96-120 hours post-induction, harvest cells for:
- qRT-PCR/Western Blot: Quantify PTEN mRNA and protein levels. Normalize to controls.
- Functional Assays: Measure dose-dependent changes in phospho-AKT levels (via Western), cell proliferation (CellTiter-Glo), and soft agar colony formation.
- Data Correlation: Plot phenotypic strength (e.g., pAKT/AKT ratio) against PTEN expression level to establish the dosage-response relationship.
Visualizations
Diagram 1: CRISPRi vs CRISPRa Core Mechanisms and Applications
Diagram 2: Pooled CRISPRi/a Screening Workflow
Diagram 3: Strategy for Titrating Gene Dosage with CRISPRa
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for CRISPRi/a Studies
| Reagent/Material | Function | Example Product/Catalog |
|---|---|---|
| dCas9-KRAB Expression Vector | Stable expression of the CRISPRi repressor machinery. | lenti dCas9-KRAB-puro (Addgene #71237) |
| dCas9-SAM Activation System | Stable expression of the optimized CRISPRa activator machinery. | pLV dCas9-SAM (Addgene #108100) |
| Genome-wide CRISPRi/a sgRNA Libraries | Pre-designed pooled libraries for loss- or gain-of-function screens. | Human Brunello CRISPRi Library (Addgene #73179), SAM sgRNA Library (Addgene #1000000078) |
| MS2-Modified sgRNA Backbone Vector | Cloning vector for sgRNAs compatible with SAM activation system. | pLX-sgRNA(MS2)-zeo (Addgene #96925) |
| Lentiviral Packaging Plasmids | For production of sgRNA or dCas9 lentiviruses. | psPAX2 (Addgene #12260), pMD2.G (Addgene #12259) |
| Inducible System Components | Allows precise temporal control of dCas9 expression (e.g., Tet-On). | pCW-Cas9 (Addgene #50661) or pTetOne-dCas9 variants |
| Next-Generation Sequencing Kit | For amplifying and preparing sgRNA barcodes from genomic DNA. | NEBNext Ultra II DNA Library Prep Kit |
| MAGeCK Software | Standard bioinformatics pipeline for analyzing CRISPR screen data. | https://sourceforge.net/p/mageck/wiki/Home/ |
| Validated Control sgRNAs | Non-targeting (negative) and essential gene targeting (positive) controls. | e.g., Non-targeting sgRNA, sgRNA targeting RPA3 |
This case study explores the application of CRISPR-Cas9-derived technologies—CRISPR knockout (CRISPRko), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa)—in the systematic validation of novel drug targets. Framed within a broader thesis comparing these modalities, we detail their mechanisms, specific use-cases in target validation, and provide a technical guide for implementation. The precision of CRISPR tools allows for the modeling of genetic perturbations that mimic drug effects, enabling high-confidence assessment of target-disease relationships before costly drug development campaigns commence.
Core Technology Comparison: CRISPRko, CRISPRi, CRISPRa
The fundamental differences between these platforms lie in the nature of the genomic perturbation and the resultant phenotypic readout, each offering unique advantages for target validation.
Mechanisms of Action
- CRISPRko: Utilizes Cas9 nuclease to create double-strand breaks (DSBs) in the coding sequence of a target gene, resulting in frameshift mutations via non-homologous end joining (NHEJ) and permanent gene knockout.
- CRISPRi: Employs a catalytically "dead" Cas9 (dCas9) fused to a transcriptional repressor domain (e.g., KRAB). The complex binds to the promoter or early exon of a target gene without cutting DNA, leading to stable, reversible transcriptional repression.
- CRISPRa: Uses dCas9 fused to transcriptional activator domains (e.g., VP64, p65, Rta). The complex is targeted to enhancer or promoter regions to upregulate gene expression, enabling gain-of-function studies.
Quantitative Comparison Table
The following table summarizes the key characteristics of each technology relevant to drug target validation.
Table 1: Functional Comparison of CRISPR Modalities in Target Validation
| Feature | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Cas9 Form | Wild-type SpCas9 (Nuclease) | dCas9 (Nuclease-dead) fused to KRAB | dCas9 fused to activator (e.g., VPR) |
| Primary Effect | Permanent gene disruption via indels | Reversible transcriptional repression | Transcriptional activation |
| Efficiency (Typical) | >80% knockout in bulk population | 70-95% repression (gene-dependent) | 5-50x activation (gene-dependent) |
| Key Application in Validation | Essentiality screens, identifying lethal targets | Modeling partial inhibition, dose-response, non-coding targets | Gain-of-function, synthetic lethality, rescuing suppressor loss |
| Major Advantage | Complete loss-of-function, mimics strong inhibitors | Tunable, reversible, fewer off-target/ploidy confounders | Models oncogene overexpression or therapeutic gene activation |
| Major Limitation | Confounds from clonal variation, essentiality bias | Repression efficiency varies by genomic context | Activation is highly context and locus-dependent |
| Best Paired With | Viability/death phenotype assays (CellTiter-Glo) | Transcriptomic (RNA-seq) & proteomic readouts | Phenotypic rescue assays, differentiation readouts |
Experimental Protocol: A Pooled CRISPRko Screen for Essential Gene Identification
The following is a detailed protocol for a genome-wide CRISPRko dropout screen to identify genes essential for cancer cell proliferation—a foundational target validation experiment.
Objective: To identify genes whose knockout leads to loss of fitness/proliferation in a specific cancer cell line.
Workflow Summary:
Detailed Protocol:
Step 1: Library Selection and Virus Production
- Select a validated genome-wide sgRNA library (e.g., Brunello library, ~4 sgRNAs/gene).
- Amplify the plasmid library and use it alongside third-generation lentiviral packaging plasmids (psPAX2, pMD2.G) to transfert HEK293T cells using PEI.
- Harvest lentiviral supernatant at 48h and 72h, concentrate via ultracentrifugation, and titer using a puromycin kill curve or qPCR-based methods.
Step 2: Cell Infection and Selection
- Plate your target cancer cells (e.g., A549) in antibiotic-free medium.
- Infect cells with the lentiviral library at an MOI of ~0.3 to ensure most cells receive only one sgRNA. Include a non-targeting control sgRNA population.
- 24h post-infection, replace medium with puromycin-containing medium (concentration determined by kill curve). Select for a minimum of 7 days until all cells in an uninfected control well are dead.
Step 3: Passaging and Harvesting
- After selection, harvest at least 20 million cells as the baseline time point (T0). This represents the initial sgRNA distribution.
- Continue culturing the remaining cells, maintaining a representation of >500 cells per sgRNA at all times (typically passaging >20 million cells per time point).
- Culture cells for approximately 14 population doublings. Harvest at least 20 million cells as the final time point (T14).
Step 4: Sequencing and Analysis
- Extract genomic DNA from T0 and T14 pellets using a maxi-prep kit.
- Perform a two-step PCR to amplify the integrated sgRNA sequences from the genomic DNA and attach Illumina sequencing adapters and sample barcodes.
- Purify the PCR product and sequence on an Illumina NextSeq (minimum 100 reads per sgRNA).
- Align reads to the sgRNA library reference file. Use computational tools like MAGeCK or CRISPRess2 to calculate the depletion (negative selection) of each sgRNA and gene between T0 and T14.
- Essential genes are identified by significant depletion (negative log2 fold-change) of multiple targeting sgRNAs (FDR < 0.05).
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for CRISPR-Based Target Validation Experiments
| Reagent / Material | Function / Purpose | Example Product/Provider |
|---|---|---|
| Validated sgRNA Library | Pre-designed, optimized pooled sgRNA sets for whole-genome or pathway-focused screens. | Brunello Human Genome-wide Library (Addgene), CRISPRi/a v2 Libraries (Addgene) |
| Lentiviral Packaging Plasmids | For producing replication-incompetent lentiviral particles to deliver CRISPR constructs. | psPAX2 (gag/pol), pMD2.G (VSV-G) (Addgene) |
| dCas9 Effector Plasmids | Express dCas9 fused to repressor (KRAB) or activator (VPR) domains for CRISPRi/a. | pLV hU6-sgRNA hUbC-dCas9-KRAB (CRISPRi), dCas9-VPR (CRISPRa) (Addgene) |
| Polybrene (Hexadimethrine Bromide) | A cationic polymer that enhances viral transduction efficiency. | Sigma-Aldrich, TR-1003 |
| Puromycin Dihydrochloride | Selective antibiotic for cells expressing resistance genes from CRISPR vectors. | Thermo Fisher Scientific, A1113803 |
| Cell Viability Assay Kit | To quantify cell proliferation/death phenotypes post-perturbation (e.g., luminescence). | CellTiter-Glo (Promega, G7571) |
| Genomic DNA Extraction Kit | For high-yield, high-purity gDNA from large cell pellets for NGS library prep. | QIAamp DNA Blood Maxi Kit (Qiagen, 51194) |
| NGS Library Prep Kit | For amplifying and barcoding sgRNA sequences from gDNA for Illumina sequencing. | NEBNext Ultra II DNA Library Prep Kit (NEB, E7645) |
| Analysis Software/Pipeline | Computationally identify significantly enriched/depleted genes from NGS count data. | MAGeCK (open source), CRISPRess2 (Broad Institute) |
Signaling Pathway Analysis Using Multi-Modal CRISPR
CRISPR technologies can deconvolve complex signaling pathways. Below is a generalized pathway highlighting how different CRISPR tools interrogate nodes for validation.
CRISPRko, CRISPRi, and CRISPRa are complementary pillars of modern genetic target validation. CRISPRko provides definitive loss-of-function evidence, CRISPRi offers nuanced, dose-responsive modeling akin to pharmacological inhibition, and CRISPRa enables gain-of-function validation and rescue studies. Integrating these approaches within a unified target validation thesis allows researchers to triangulate high-confidence therapeutic targets, deconvolve complex signaling networks, and ultimately derisk downstream drug development. The continued refinement of specificity, efficiency, and delivery for each modality will further solidify CRISPR's role as an indispensable tool in the translational research pipeline.
Maximizing Efficiency and Specificity: Troubleshooting Common Pitfalls
Within the paradigm of CRISPR-based transcriptional modulation, the core technologies of CRISPR knockout (CRISPRko), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa) present distinct molecular mechanisms and, consequently, unique profiles of off-target effects. This guide details the specific challenges for each modality, providing a technical framework for their identification and mitigation, essential for therapeutic and research applications.
Defining Off-Target Effects by Modality
CRISPRko (Nuclease-Mediated Gene Knockout)
Off-target effects arise primarily from Cas9 nuclease activity at genomic loci with sequences similar to the intended sgRNA target. Mismatches, particularly in the seed region proximal to the PAM, can be tolerated, leading to double-strand breaks (DSBs) at unintended sites.
Primary Challenge: Indels and chromosomal rearrangements at off-target sites can disrupt functional genes or regulatory elements, confounding phenotypic readouts and posing significant safety risks.
CRISPRi (dCas9-Mediated Transcriptional Repression)
CRISPRi utilizes a catalytically dead Cas9 (dCas9) fused to a repressive domain (e.g., KRAB) to block transcription. Off-targets are defined by dCas9 binding at similar sequences without cleavage.
Primary Challenge: Epigenetic silencing and transcriptional repression at off-target loci can lead to gene expression dysregulation. These effects are reversible but can produce misleading results in genetic screens or modulate pathways unintentionally.
CRISPRa (dCas9-Mediated Transcriptional Activation)
CRISPRa employs dCas9 fused to transcriptional activators (e.g., VPR, p65AD) to upregulate gene expression. Like CRISPRi, off-targets stem from dCas9 binding.
Primary Challenge: Ectopic activation of genes, including proto-oncogenes or genes influencing cellular state, can create false positives in screens and potential oncogenic hazards. The risk of over-activating nearby genes via "looping" or enhancer effects is also present.
Quantitative Comparison of Off-Target Profiles
Table 1: Comparative Analysis of Off-Target Effects
| Feature | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Primary Mechanism | Cas9-induced DSB & NHEJ/HDR | dCas9-KRAB binding & chromatin silencing | dCas9-activator binding & chromatin opening |
| Nature of Effect | Permanent genetic deletion/insertion | Reversible transcriptional repression | Reversible transcriptional activation |
| Key Off-Target Risk | Indels at genomic sites with sequence homology | Silencing of genes with similar regulatory regions | Activation of genes with similar regulatory regions |
| Typical Detection Method | Whole-genome sequencing (WGS), GUIDE-seq, CIRCLE-seq | ChIP-seq (for dCas9 binding), RNA-seq | ChIP-seq (for dCas9/activator), RNA-seq |
| Reported Off-Target Rate (Range)* | 0-50+ sites (highly sgRNA-dependent) | Binding: 10-100s sites; Functional: <5% of bound | Binding: 10-100s sites; Functional: <5% of bound |
| Potential for Aneuploidy | High (due to DSBs) | Very Low | Very Low |
*Rates are illustrative and highly dependent on sgRNA design, delivery, and cell type.
Table 2: Common Mitigation Strategies
| Strategy | Effectiveness for Ko | Effectiveness for i/a |
|---|---|---|
| High-Fidelity Cas9 Variants (e.g., SpCas9-HF1) | High (Reduces nuclease activity at mismatches) | Moderate (Improves binding specificity) |
| Truncated sgRNAs (tru-gRNAs) | Moderate | High (Reduces binding energy, increasing specificity) |
| Paired Nickases (e.g., Cas9n) | High (Requires two adjacent off-targets) | Not Applicable |
| Optimal sgRNA Design (Algorithmic) | Critical | Critical |
| Promoter/Enhancer Mapping | Less Relevant | High (Avoids targeting in dense regulatory regions) |
Experimental Protocols for Off-Target Assessment
Protocol: Genome-Wide Off-Target Detection for CRISPRko (GUIDE-seq)
Principle: Captures double-strand breaks genome-wide by integrating a double-stranded oligodeoxynucleotide (dsODN) tag.
Method:
- Transfection: Co-deliver Cas9:sgRNA RNP and the dsODN GUIDE-seq tag into target cells.
- Genomic DNA Extraction: Harvest cells 72 hours post-transfection. Extract high-molecular-weight gDNA.
- Library Preparation: Shear gDNA, enrich for tag-integrated fragments via PCR, and prepare sequencing library.
- Sequencing & Analysis: Perform NGS. Use the GUIDE-seq computational pipeline to align reads and identify off-target integration sites.
Protocol: Binding Specificity Assessment for CRISPRi/a (dCas9 ChIP-seq)
Principle: Maps genome-wide binding sites of dCas9-repressor/activator fusions.
Method:
- Cell Line Generation: Stably express dCas9-effector fusion (KRAB or VPR) and sgRNA of interest.
- Crosslinking & Shearing: Fix cells with formaldehyde. Sonicate chromatin to ~200-500 bp fragments.
- Immunoprecipitation: Use an antibody against the epitope-tagged dCas9 (e.g., FLAG, HA) to pull down bound DNA fragments.
- Library Prep & Sequencing: Reverse crosslinks, purify DNA, and prepare sequencing library. Sequence.
- Analysis: Align reads, call peaks (e.g., with MACS2). Compare peak locations to the intended target and potential off-target sequences.
Protocol: Transcriptional Off-Target Validation (RNA-seq)
Principle: Quantifies genome-wide expression changes following CRISPRi/a perturbation.
Method:
- Perturbation: Perform CRISPRi or CRISPRa experiment alongside non-targeting sgRNA control.
- RNA Harvest: Extract total RNA 48-96 hours post-perturbation. Ensure high RIN.
- Library Preparation: Deplete rRNA, perform poly-A selection, and construct strand-specific RNA-seq libraries.
- Sequencing & Analysis: Sequence to sufficient depth (≥30M reads). Align to reference genome, quantify gene expression (e.g., with Salmon). Use DESeq2 to identify differentially expressed genes (FDR < 0.05). Exclude the targeted gene to focus on off-target transcriptional changes.
Visualizing Mechanisms and Workflows
Diagram Title: Core Off-Target Mechanisms for CRISPRko, i, and a
Diagram Title: Off-Target Assessment Workflow for Ko vs i/a
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Off-Target Analysis
| Item | Function | Example Product/Catalog |
|---|---|---|
| High-Fidelity Cas9 Nuclease | Reduces off-target cleavage for CRISPRko. | SpCas9-HF1 (Integrated DNA Technologies) |
| dCas9-KRAB Expression Plasmid | Enables stable CRISPRi for binding/expression assays. | lenti dCas9-KRAB-blast (Addgene #89567) |
| dCas9-VPR Expression Plasmid | Enables stable CRISPRa for binding/expression assays. | pHAGE dCas9-VPR (Addgene #63810) |
| GUIDE-seq dsODN Tag | Double-stranded tag for genome-wide DSB detection. | GUIDE-seq Oligo (Integrated DNA Technologies) |
| Anti-FLAG M2 Magnetic Beads | For immunoprecipitation in dCas9 ChIP-seq experiments. | Sigma-Aldrich M8823 |
| Strand-Specific RNA-seq Kit | Prepares libraries for transcriptome analysis of CRISPRi/a effects. | Illumina Stranded Total RNA Prep |
| Next-Generation Sequencing Service | Provides deep sequencing for GUIDE-seq, ChIP-seq, RNA-seq. | Illumina NovaSeq, MiSeq |
| CRISPR Design Software | Algorithms to predict on-target efficiency and potential off-targets. | CRISPick (Broad Institute), ChopChop |
| Off-Target Analysis Pipeline | Bioinformatics tools for processing sequencing data. | GUIDE-seq (Magoc et al.), MACS2 (for ChIP-seq), DESeq2 (for RNA-seq) |
Optimizing sgRNA Design and Delivery for Each Modality
1. Introduction The selection of a CRISPR interference modality—CRISPR knockout (CRISPRko), CRISPR interference (CRISPRi), or CRISPR activation (CRISPRa)—dictates fundamental requirements for single guide RNA (sgRNA) design and delivery. This guide details optimized strategies for each approach within the context of comparative functional genomics and therapeutic development.
2. Modality-Specific sgRNA Design Principles Design parameters diverge significantly based on the intended genomic perturbation. Key quantitative considerations are summarized below.
Table 1: Core sgRNA Design Parameters by Modality
| Parameter | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Target Region | Early coding exons (esp. 2nd+), essential functional domains. | Proximal to Transcription Start Site (TSS), -50 to +300 bp relative to TSS. | Proximal to TSS, -400 to -50 bp upstream of TSS. |
| Optimal GC% | 40-60% | 30-70% (broader tolerance). | 30-70% (broader tolerance). |
| Specificity | Minimize off-targets via predictive algorithms (e.g., CFD score). | Tolerates more off-targets; repression is reversible & titratable. | Critical for minimizing aberrant activation; high specificity required. |
| PAM Requirement | Standard SpCas9 NGG. | dCas9 or dCas9-KRAB uses SpCas9 NGG. | dCas9-VPR or dCas9-p300 uses SpCas9 NGG. |
| Key Algorithm | Doench '16, CHOPCHOP, MIT/Broad sgRNA Designer. | CRISPRi design tools (e.g., from Weissman/St. Jude labs). | CRISPRa design tools (e.g., from Weissman/St. Jude labs). |
| Typical Efficacy | ~80-95% indel formation (varies by locus). | ~70-90% gene repression (knockdown). | ~2-10x gene activation (highly variable by locus). |
3. Delivery Strategies Optimized for Modality Effective delivery is contingent on payload size, duration of expression, and cellular context.
Table 2: Delivery Vehicles for CRISPR Modalities
| Delivery Method | Max Capacity | Best For | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Lentivirus (LV) | ~8 kb | CRISPRi, CRISPRa, pooled screens. | Stable genomic integration, long-term expression. | Random insertional risk, size constraints. |
| Adeno-Associated Virus (AAV) | ~4.7 kb | CRISPRko (SaCas9), in vivo delivery. | Low immunogenicity, high in vivo transduction. | Very limited cargo size; requires split systems for SpCas9. |
| Electroporation (RNP) | N/A (direct delivery) | CRISPRko in primary/immune cells, ex vivo therapy. | High efficiency, rapid degradation reduces off-targets. | Transient effect, not suitable for stable repression/activation. |
| Lipid Nanoparticles (LNP) | Variable (mRNA/sgRNA) | CRISPRko, transient CRISPRi/a, in vivo systemic delivery. | Clinical relevance, scalable, high efficiency in vivo. | Transient expression, potential immunogenicity. |
4. Detailed Experimental Protocols
Protocol 4.1: Design and Cloning of a CRISPRi/a sgRNA Library for a Genome-Wide Screen Objective: To construct a lentiviral sgRNA library targeting gene promoters for a CRISPRi or CRISPRa screen. Materials: Oligo pool (designed per Table 1), lenti-Guide-Puro or lenti-sgRNA(MS2)_zeo backbone, NEBuilder HiFi DNA Assembly Master Mix, Endura electrocompetent cells, QIAprep Spin Miniprep Kit, LB agar plates with appropriate antibiotic. Procedure:
- Oligo Pool Design: Using a validated CRISPRi/a design rule set, generate a 20mer sgRNA sequence for each target. Add flanking cloning sequences (e.g., for BsmBI sites).
- Annealing & Phosphorylation: Resuspend oligo pool, anneal, and phosphorylate using T4 PNK.
- Backbone Digestion: Digest the lentiviral backbone plasmid with BsmBI-v2 for 2 hours at 37°C. Gel-purify the linearized vector.
- Golden Gate Assembly: Perform a Golden Gate assembly using BsmBI and T7 DNA Ligase, cycling between 37°C (ligation) and 16°C (digestion) for 30 cycles.
- Transformation: Transform the assembled product into Endura electrocompetent cells via electroporation. Plate on large 245 x 245 mm LB agar plates to ensure >200x library representation.
- Plasmid Harvest: Incubate for 16 hours. Scrape all colonies for maxiprep plasmid DNA purification. Validate library complexity by deep sequencing.
Protocol 4.2: Evaluating CRISPRko vs. CRISPRi Efficiency via Flow Cytometry Objective: To compare gene knockout (indel) efficiency vs. transcriptional knockdown efficiency for a target gene. Materials: HEK293T cells, Lipofectamine 3000, plasmids: lentiCas9-Blast, lenti-sgRNA(CRISPRko), lenti-dCas9-KRAB-Blast, lenti-sgRNA(CRISPRi) targeting the same locus, antibody for target protein (if available), flow cytometer. Procedure:
- Cell Line Generation: Generate stable Cas9 and dCas9-KRAB cell lines via lentiviral transduction and blasticidin selection.
- sgRNA Transduction: Transduce both cell lines in parallel with lentivirus carrying the target sgRNA (or non-targeting control). Apply puromycin selection.
- Harvest and Stain: 7 days post-transduction, harvest cells. For a surface protein target, stain with a fluorescently conjugated antibody. For an intracellular target, perform fixation/permeabilization prior to staining.
- Flow Analysis: Acquire data on a flow cytometer. For CRISPRko, calculate the percentage of protein-negative cells. For CRISPRi, calculate the mean fluorescence intensity (MFI) shift relative to control.
- Validation: For CRISPRko, isolate genomic DNA from edited pools and perform T7 Endonuclease I assay or next-generation sequencing (NGS) of the target locus to quantify indels.
5. Visualizing Workflows and Pathways
Title: Decision Flow for CRISPR Modality Optimization
Title: Core Protein Complexes and Outcomes by Modality
6. The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for CRISPR Modality Research
| Reagent / Material | Supplier Examples | Function |
|---|---|---|
| lentiCas9-Blast & lentiGuide-Puro | Addgene #52962, #52963 | Standardized plasmids for stable CRISPRko cell line generation and sgRNA expression. |
| lenti-dCas9-KRAB-Blast & lenti-sgRNA(MS2)_zeo | Addgene #89567, #89308 | Essential for CRISPRi; dCas9 fused to KRAB repressor, with compatible sgRNA scaffold. |
| lenti-dCas9-VPR-Blast | Addgene #63798 | Essential for CRISPRa; dCas9 fused to VPR tripartite activator. |
| High-Efficiency sgRNA Cloning Kit | e.g., ToolGen, Synthego | Streamlines BsmBI-based cloning of sgRNA libraries into lentiviral backbones. |
| Endura Electrocompetent Cells | Lucigen | High-transformation-efficiency bacteria for large, complex library cloning. |
| Lipofectamine 3000 | Thermo Fisher | High-efficiency transfection reagent for plasmid delivery in vitro. |
| Puromycin Dihydrochloride | Sigma-Aldrich, Thermo Fisher | Selection antibiotic for cells transduced with puromycin-resistant sgRNA vectors. |
| T7 Endonuclease I | NEB | Enzyme for detecting Cas9-induced indels via mismatch cleavage assay. |
| NEBuilder HiFi DNA Assembly Master Mix | NEB | For seamless assembly of larger CRISPR components (e.g., dCas9 fusions). |
Boosting Knockdown Efficiency in CRISPRi and Activation in CRISPRa
Within the broader thesis comparing CRISPRko (knockout), CRISPRi (interference), and CRISPRa (activation), optimizing efficiency is paramount for functional genomics and therapeutic discovery. CRISPRi and CRISPRa offer reversible, tunable transcriptional modulation, unlike permanent DNA cleavage by CRISPRko. This guide details technical strategies to maximize knockdown and activation efficacy.
Quantitative Comparison of CRISPRko, CRISPRi, and CRISPRa
Table 1: Core Characteristics and Performance Metrics of CRISPR Modalities
| Parameter | CRISPRko (Knockout) | CRISPRi (Interference) | CRISPRa (Activation) |
|---|---|---|---|
| Catalytic Core | Wild-type Cas9 (SpCas9) | Deactivated Cas9 (dCas9) fused to repressor domains (e.g., KRAB) | Deactivated Cas9 (dCas9) fused to activator domains (e.g., VPR, SAM) |
| Primary Mechanism | Creates double-strand breaks (DSBs) leading to indel mutations. | Blocks transcription initiation/elongation; recruits chromatin compactors. | Recruits transcriptional machinery and opens chromatin. |
| Typical Knockdown/Activation Efficiency | Near 100% protein loss (for frameshift indels). | Typically 70-95% mRNA knockdown. | Often 2-10+ fold mRNA upregulation; varies widely by target. |
| Kinetics | Permanent; rapid protein depletion post-repair. | Reversible; effects manifest within 24-48h. | Reversible; effects manifest within 24-72h. |
| Key Influencing Factors | HDR/NHEJ repair balance; sgRNA cutting efficiency. | sgRNA proximity to TSS; chromatin state; repressor strength. | sgRNA proximity to TSS/Enhancer; chromatin state; activator system. |
| Primary Applications | Essential gene studies; creating knockout cell lines. | Gene function studies; modeling hypomorphs; synthetic circuits. | Gain-of-function studies; cellular reprogramming; therapeutic upregulation. |
Table 2: Strategies to Boost Efficiency in CRISPRi and CRISPRa
| Strategy | CRISPRi Application | CRISPRa Application | Quantitative Impact |
|---|---|---|---|
| Multiplexing sgRNAs | Target multiple sites near TSS for synergistic repression. | Target multiple enhancer regions or promoter-proximal sites. | Can increase repression from ~80% to >95% or activation from 5-fold to 20+ fold. |
| Optimized Effector Domains | Use KRAB-MeCP2 fusions vs. KRAB alone. | Use VPR (VP64-p65-Rta) or SAM (Synergistic Activation Mediator) systems. | Strong domains can improve efficacy by 1.5-3x over base systems. |
| Chromatin-Modifying Fusions | Fuse dCas9 to DNMT3A for DNA methylation. | Fuse dCas9 to p300 core for histone acetylation (H3K27ac). | Epigenetic silencing can yield >90% repression; p300 can boost activation ~2-5x in heterochromatin. |
| sgRNA Positioning | Place sgRNA -50 to +300 bp relative to TSS. | For SAM, place sgRNA -200 to -50 bp upstream of TSS. | Optimal positioning can mean 10x difference in activation/repression output. |
| Promoter/Enhancer Tiling | Systematic screening of sgRNAs across promoter region. | tiling across putative enhancer regions (e.g., via ATAC-seq peaks). | Identifies "hot spots" where efficacy can be 5x higher than average. |
| MS2/PP7 RNA Loop Engineering | Incorporate MS2 loops to recruit MCP-KRAB for added repression. | Core of SAM system: MS2 loops recruit MCP-p65-HSF1 activators. | Enables recruitment of multiple effectors, boosting activation (SAM) 10-100x over dCas9-VP64. |
Experimental Protocols for Efficiency Optimization
Protocol 2.1: High-Efficiency CRISPRi Knockdown Workflow
Objective: Achieve >90% transcriptional knockdown of a target gene in HEK293T cells.
Materials: See "Scientist's Toolkit" below. Procedure:
- sgRNA Design & Cloning:
- Design 3-5 sgRNAs targeting the region from -50 to +300 bp relative to the annotated Transcription Start Site (TSS). Use algorithms like CRISPRi/v2 or Rule Set 2.0 for prediction.
- Clone sgRNA sequences into a lentiviral dCas9-KRAB-MeCP2 expression vector (e.g., pLV hU6-sgRNA hUbC-dCas9-KRAB-MeCP2) via BsmBI Golden Gate assembly.
- Lentivirus Production:
- Co-transfect HEK293T producer cells with the sgRNA vector, psPAX2 (packaging), and pMD2.G (VSV-G envelope) plasmids using a PEI transfection reagent.
- Harvest virus-containing supernatant at 48 and 72 hours post-transfection, concentrate via ultracentrifugation.
- Cell Line Generation & Validation:
- Transduce target cells with the lentivirus in the presence of 8 µg/mL polybrene. Spinfect at 1000 x g for 1 hour at 32°C to enhance infection.
- Select stable pools with 2 µg/mL puromycin for 7 days.
- Validate knockdown by RT-qPCR 7 days post-selection:
- Extract total RNA, synthesize cDNA.
- Perform qPCR with TaqMan probes for the target gene and a reference gene (e.g., GAPDH).
- Calculate % knockdown via the 2^(-ΔΔCt) method. Aim for pools with >85% knockdown. For maximal effect, single-cell clone isolation and screening may be required.
Protocol 2.2: Multiplexed CRISPRa for Robust Gene Activation
Objective: Achieve >20-fold gene activation using the SAM system.
Materials: See "Scientist's Toolkit" below. Procedure:
- Multiplex sgRNA Vector Assembly:
- Design 3 sgRNAs targeting positions between -200 to -50 bp upstream of the TSS. Include an additional sgRNA targeting a known positive control gene locus (e.g., MYOD1).
- Use a multiplexed sgRNA scaffold vector (e.g., pLenti-sgRNA-MS2-Puro containing two BsmBI sites for array cloning) to assemble a tandem sgRNA expression cassette via Golden Gate assembly.
- Co-delivery of SAM Components:
- The SAM system requires three components: dCas9-VP64, MS2-p65-HSF1 (activator), and the sgRNA-MS2.
- Method A (Lentiviral): Co-transduce target cells with lentiviruses for dCas9-VP64 and MS2-p65-HSF1, then with the multiplex sgRNA virus. Select with appropriate antibiotics (e.g., Blasticidin and Hygromycin).
- Method B (Transient Transfection): For rapid testing, co-transfect a stable dCas9-VP64 cell line with plasmids expressing MS2-p65-HSF1 and the multiplex sgRNA vector using Lipofectamine 3000.
- Activation Analysis:
- Harvest cells 72 hours post-transduction/transfection.
- Perform RT-qPCR as in Protocol 2.1 to assess mRNA levels.
- Normalize target gene expression to the negative control (non-targeting sgRNA) and the positive control (MYOD1 activation). Activation fold-change is calculated as 2^(ΔΔCt).
- For protein-level validation, perform Western blot 5-7 days post-transduction.
Visualizations
Title: CRISPRi High-Efficiency Workflow
Title: CRISPRa SAM System Mechanism
The Scientist's Toolkit
Table 3: Essential Research Reagents for Optimizing CRISPRi/a
| Item | Function in CRISPRi/a | Example Product/Catalog # (Representative) |
|---|---|---|
| dCas9 Effector Plasmids | Expresses the core dCas9 fused to repressor or activator domains. | CRISPRi: pHR-SFFV-dCas9-BFP-KRAB (Addgene #46911). CRISPRa: dCas9-VP64_Blast (Addgene #61425). |
| sgRNA Cloning Backbone | Vector for expressing sgRNA, often with MS2 loops for CRISPRa. | lentiGuide-Puro (Addgene #52963) / lenti-sgRNA-MS2-Puro (for SAM, Addgene #73797). |
| Lentiviral Packaging Mix | For producing safe, high-titer lentivirus to deliver constructs. | psPAX2 (Addgene #12260) & pMD2.G (Addgene #12259) or commercial kits (e.g., Lenti-X from Takara). |
| Polycation Transfection Reagent | Enhances lentiviral transduction efficiency in difficult cells. | Polybrene (Hexadimethrine bromide) or LentiBoost (Sirion Biotech). |
| Epigenetic Modifier Fusions | Advanced effectors for persistent silencing or strong activation. | dCas9-DNMT3A (for silencing, Addgene #71666); dCas9-p300 Core (for activation, Addgene #61357). |
| Validated Positive Control sgRNA | Essential for normalizing and benchmarking system performance. | Non-targeting control sgRNA; CRISPRa: sgRNA targeting MYOD1 promoter. |
| RT-qPCR Kit with Probes | Gold-standard for precise quantification of mRNA knockdown/activation. | TaqMan RNA-to-Ct 1-Step Kit or equivalent. |
| Next-Gen Sequencing Library Prep Kit | For genome-wide CRISPRi/a screen readout and off-target analysis. | Illumina Nextera XT or similar for sgRNA library amplification. |
Mitigating Toxicity and Adaptive Responses in Long-Term Experiments
The pursuit of precise, long-term genetic perturbations using CRISPR technologies—CRISPR knockout (CRISPRko), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa)—is fundamental to functional genomics and therapeutic target validation. However, sustained manipulation of cellular systems often triggers toxicity and adaptive responses that confound experimental outcomes. This guide addresses these challenges within the specific context of differentiating the durable effects of complete gene ablation (ko), transcriptional silencing (i), and overexpression (a). Mitigating unintended consequences is critical for deriving accurate biological insights, especially in prolonged assays such as long-term culture, drug resistance studies, and in vivo model development.
Core Challenges: Toxicity and Adaptation in Sustained Perturbations
- On-Target Toxicity: Essential gene knockout or severe repression can induce cell death or cell cycle arrest, leading to selective pressure and outgrowth of escapers.
- Off-Target Effects: Persistent nuclease (for ko) or dCas9-effector (for i/a) activity increases the risk of DNA damage responses or transcriptional noise.
- Adaptive Responses: Cells may activate compensatory pathways, epigenetic remodeling, or feedback loops to counteract the perturbation over time, masking the primary phenotype.
- Technological Drift: For CRISPRi/a, sustained binding may lead to squelching of dCas9-effectors or changes in chromatin state, reducing efficacy.
Strategic Framework for Mitigation
A multi-layered approach is required to ensure robustness.
Experimental Design & Controls
- Inducible Systems: Use doxycycline- or small-molecule-inducible dCas9 systems to control the timing and duration of perturbation, allowing recovery periods.
- Multi-Guide Designs: Employ multiple gRNAs per target to distinguish consistent on-target effects from guide-specific artifacts.
- Scrambled/Non-Targeting Controls: Essential for benchmarking background rates of adaptation.
- Parallel ko/i/a Comparisons: Directly comparing all three modalities for the same target gene can isolate effects specific to perturbation type versus shared adaptive responses.
Protocol Optimization
- Titration of Effector Expression: Lower dCas9-effector levels (e.g., using weaker promoters) can minimize toxicity while maintaining efficacy.
- Kinetic Monitoring: Implement longitudinal sampling (e.g., Day 3, 7, 14, 30) for transcriptomic (RNA-seq) and phenotypic analysis to track evolution of responses.
- Clonal vs. Polyclonal Analysis: While polyclonal populations capture heterogeneous adaptations, isolating and profiling multiple single-cell clones can reveal distinct escape mechanisms.
Quantitative Comparison of Toxicity Profiles
Table 1: Comparative Toxicity and Adaptation Risks in Long-Term CRISPR Perturbations
| Perturbation Type | Core Mechanism | Primary Toxicity Risk | Common Adaptive Responses | Typical Onset of Adaptation | Key Mitigation Strategy |
|---|---|---|---|---|---|
| CRISPRko | NHEJ/MMEJ-induced frameshift mutations. | Loss of essential gene function; p53-mediated DNA damage response from double-strand breaks. | Selection for in-frame edits or heterozygous knockout; amplification of paralogous genes. | Days 7-14 in continuous culture. | Use of inducible Cas9; parallel use of CRISPRi as a conditional mimic. |
| CRISPRi | dCas9-KRAB-mediated transcriptional repression. | Silencing of essential genes; potential squelching of endogenous KRAB pathways. | Upregulation of compensatory genes via feedback loops; chromatin remodeling. | Days 10-21. | Titration of dCas9-KRAB expression; pulsed repression cycles. |
| CRISPRa | dCas9-VPR/p300-mediated transcriptional activation. | Overexpression-induced proteotoxic stress; disruption of regulatory networks. | Downregulation of upstream activators; silencing of the activated locus via heterochromatin. | Days 5-14. | Use of weaker activation domains (e.g., SunTag-VP64); inducible systems. |
Table 2: Example Reagent Solutions for Mitigation Experiments
| Reagent/Catalog Tool | Primary Function in Mitigation | Example Vendor(s) |
|---|---|---|
| Doxycycline-inducible dCas9 vectors | Enables temporal control of perturbation onset/duration. | Addgene, Thermo Fisher Scientific |
| Blastidin/Puromycin selection markers | Maintains stable expression of CRISPR constructs over long-term culture. | Sigma-Aldrich, Invivogen |
| CRISPRko/i/a Benchmark Library | Pre-designed libraries for direct comparison of perturbation types for the same gene set. | Synthego, Dharmacon |
| Cell Titer-Glo / Incucyte | Longitudinal, non-invasive monitoring of cell viability/proliferation. | Promega, Sartorius |
| Single-cell RNA-seq kits (10x Genomics) | Profiling heterogeneous adaptive responses in a polyclonal population. | 10x Genomics, Parse Biosciences |
Detailed Experimental Protocols
Protocol 1: Longitudinal Tracking of Perturbation Efficacy and Viability
Objective: To quantify the stability of gene perturbation and concurrent cell fitness over 4 weeks. Materials: Inducible CRISPRko/i/a cell line, appropriate selection antibiotics, inducer (e.g., doxycycline), qPCR reagents, flow cytometer. Steps:
- Seed polyclonal population and induce perturbation at Day 0. Maintain parallel non-induced controls.
- At weekly intervals (Days 7, 14, 21, 28), harvest an aliquot of cells.
- For mRNA: Extract RNA, perform RT-qPCR for target gene and housekeeping/compensatory genes.
- For Viability: Perform Cell Titer-Glo assay and count total cell numbers.
- For Protein: Harvest protein for Western blot of target (if antibodies available).
- Passage cells continuously, maintaining selection and inducer.
- Analysis: Plot relative target expression (ko/i/a) and normalized cell viability versus time. A drop in perturbation efficacy concurrent with a recovery in viability suggests adaptive outgrowth.
Protocol 2: Single-Cell Clone Isolation & Characterization to Decipher Adaptation
Objective: To isolate and molecularly profile individual clones that survive long-term essential gene perturbation. Materials: 96-well plates, cloning discs, puromycin, genomic DNA extraction kit, PCR primers for target locus, NGS library prep kit. Steps:
- After 3-4 weeks of selection under perturbation, isolate single cells by FACS or limiting dilution into 96-well plates.
- Expand clones for 2-3 weeks.
- Genotype each clone:
- For CRISPRko: PCR-amplify the target locus from gDNA; sequence via Sanger or NGS to characterize mutations.
- For CRISPRi/a: Perform RT-qPCR to assess residual repression or activation.
- For clones with unexpected genotypes/phenotypes (e.g., viable despite targeting an essential gene), perform RNA-seq to identify transcriptional adaptations.
Visualizing Pathways and Workflows
Diagram Title: Workflow for Identifying Adaptive Responses
Diagram Title: Common Adaptive Pathways to CRISPRko and CRISPRi
Within the broader thesis comparing CRISPR knockout (CRISPRko), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa), robust validation of intended perturbations is paramount. Each modality—complete gene ablation, transcriptional repression, or targeted upregulation—creates distinct molecular outcomes. Validating these outcomes with appropriate readout technologies (qPCR, RNA-seq, proteomics) ensures accurate interpretation of phenotypic data and is critical for research and drug development.
Core Validation Readouts: Technical Comparison
| Readout Method | Throughput | Quantitative Precision | Dynamic Range | Primary Application in Perturbation Validation | Key Limitation |
|---|---|---|---|---|---|
| qPCR | Low to Medium | High (exact copy number) | ~7-8 logs | Targeted mRNA-level confirmation of CRISPRi/a efficiency; validation of RNA-seq hits. | Requires prior knowledge; limited multiplexing. |
| RNA-seq (Bulk) | High | Moderate (counts normalized) | >5 logs | Genome-wide transcriptome changes for all modalities; off-target effect screening. | Cost; data complexity; indirect protein inference. |
| RNA-seq (Single-cell) | Very High | Lower per cell | >4 logs | Resolving cell-to-cell heterogeneity in perturbation response in pooled screens. | Highest cost; complex bioinformatics. |
| Mass Spec Proteomics | Medium to High | Moderate to High | ~4-5 logs | Direct measurement of protein abundance post-perturbation; gold standard for KO validation. | Lower sensitivity than nucleic acid methods; higher sample requirement. |
Detailed Methodologies for Key Experiments
Validation of CRISPRi/a Efficiency via RT-qPCR
Protocol: Following transduction/transfection of CRISPR guide RNAs and dCas9-KRAB (CRISPRi) or dCas9-VPR (CRISPRa), cells are harvested after a defined period (typically 72-96 hours).
- RNA Extraction: Use a column-based kit with on-column DNase I treatment.
- cDNA Synthesis: Use 500 ng - 1 µg total RNA with random hexamers and a reverse transcriptase enzyme.
- qPCR Setup: Design 3-4 primer pairs targeting the gene(s) of interest and 2-3 validated reference genes (e.g., GAPDH, ACTB, HPRT1). Use a SYBR Green or TaqMan master mix. Run in technical triplicates.
- Data Analysis: Calculate ∆∆Cq values. For CRISPRi, expect >70% knockdown. For CRISPRa, fold-changes can vary widely (often 5-50x) depending on the endogenous locus.
Genome-wide Validation via Bulk RNA-seq
Protocol: For comprehensive assessment of on-target and off-target effects across CRISPRko/i/a.
- Library Preparation: Use 500 ng - 1 µg of high-quality total RNA (RIN > 8.5). Employ a poly-A selection kit for mRNA enrichment. Use a stranded library prep kit (e.g., Illumina TruSeq) to preserve strand information.
- Sequencing: Aim for 25-40 million paired-end reads (e.g., 2x150 bp) per sample on an Illumina platform.
- Bioinformatic Analysis:
- Alignment: Map reads to the human/mouse reference genome (e.g., GRCh38) using STAR or HISAT2.
- Quantification: Generate gene-level counts using featureCounts or similar.
- Differential Expression: Use DESeq2 or edgeR to compare perturbation to control. For CRISPRko, expect nonsense-mediated decay (NMD) signatures. For CRISPRi/a, expect specific down/up-regulation without NMD.
Proteomic Validation via LC-MS/MS
Protocol: Essential for confirming loss-of-protein in CRISPRko and measuring downstream effects.
- Sample Preparation: Harvest cells, lyse in RIPA buffer with protease inhibitors. Reduce, alkylate, and digest proteins with trypsin.
- Peptide Cleanup: Desalt using C18 solid-phase extraction tips or columns.
- LC-MS/MS Analysis: Use a nanoflow HPLC system coupled to a high-resolution tandem mass spectrometer (e.g., Orbitrap).
- Data Processing: Identify and quantify peptides using software (e.g., MaxQuant, Proteome Discoverer) against a species-specific protein database. Normalize label-free intensities (LFQ) across samples.
Visualizing Validation Workflows and Pathways
Title: Multi-Omics Validation Workflow for CRISPR Perturbations
Title: Molecular Outcomes of CRISPRko, i, and a Modalities
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function in Perturbation Validation | Example Vendor/Product |
|---|---|---|
| DNase I, RNase-free | Eliminates genomic DNA contamination during RNA isolation for qPCR/RNA-seq. | Thermo Fisher, Qiagen |
| High-Capacity cDNA Reverse Transcription Kit | Converts purified RNA into stable cDNA for downstream qPCR analysis. | Applied Biosystems |
| SYBR Green or TaqMan Master Mix | Fluorescent chemistry for quantitative real-time PCR amplification and detection. | Bio-Rad, Thermo Fisher |
| TruSeq Stranded mRNA Library Prep Kit | Prepares strand-specific, indexed RNA-seq libraries for Illumina sequencing. | Illumina |
| Chromium Next GEM Single Cell Kit | Enables droplet-based single-cell partitioning and barcoding for scRNA-seq. | 10x Genomics |
| RIPA Lysis Buffer | Comprehensive cell lysis buffer for total protein extraction prior to proteomics. | MilliporeSigma |
| Trypsin, Sequencing Grade | High-purity protease for digesting proteins into peptides for LC-MS/MS analysis. | Promega |
| C18 Desalting Tips (StageTips) | Microscale cleanup and desalting of peptide samples prior to mass spectrometry. | Thermo Fisher |
| Reference RNA (ERCC Spike-in Mix) | Exogenous RNA controls added to samples for normalization in RNA-seq. | Thermo Fisher |
| CRISPR Modality-Specific Positive Control Guides | Validated sgRNAs known to efficiently repress (i) or activate (a) a housekeeping gene. | Synthego, Horizon Discovery |
Key Controls and Reagents for Ensuring Experimental Rigor
Within the context of comparative functional genomics research—specifically, studies delineating the differences between CRISPR knockout (CRISPRko), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa)—ensuring experimental rigor is paramount. The choice of control experiments and validation reagents directly impacts the reliability and interpretability of data, especially when drawing conclusions about the relative efficacy, specificity, and phenotypic outcomes of these distinct perturbation modalities. This guide details the essential controls and core reagents necessary for robust experimental design.
I. Core Controls for CRISPR Perturbation Experiments
Appropriate controls isolate the specific effects of the genetic perturbation from technical artifacts. The table below summarizes the key control types.
Table 1: Essential Experimental Controls for CRISPRko/i/a Studies
| Control Type | Purpose in CRISPRko | Purpose in CRISPRi | Purpose in CRISPRa | Example |
|---|---|---|---|---|
| Non-Targeting Guide Control | Distinguish on-target effects from cellular responses to Cas9 cutting/DNA damage. | Distinguish dCas9-binding/repression from specific guide-mediated effects. | Distinguish dCas9-binding/activation from specific guide-mediated effects. | Guide RNA targeting a safe-harbor locus (e.g., AAVS1) or scrambled sequence. |
| Targeting Efficiency Control | Confirm loss-of-function via indels (e.g., T7E1 assay, NGS). | Confirm transcriptional repression via qRT-PCR. | Confirm transcriptional activation via qRT-PCR. | Guide with known high efficiency against a positive control gene (e.g., HPRT1 knockdown). |
| Component Controls | Verify Cas9 dependency. | Verify dCas9-KRAB or dCas9-VPR dependency. | Verify dCas9-VPR or dCas9-p300 dependency. | Cells transfected with guide RNA but lacking the Cas9/dCas9 effector. |
| Phenotype Rescue Control | Confirm on-target effect by reintroducing cDNA resistant to the guide. | Confirm on-target effect by removing doxycycline (for inducible systems) or using orthogonal repression. | Confirm on-target effect by removing doxycycline or using orthogonal activation. | Expression of a guide-resistant wild-type cDNA for a CRISPRko hit. |
| Multi-Guide Concordance | Rule out off-target effects; phenotype should be consistent across ≥2 guides per gene. | Rule off-target repression; phenotype should be consistent across ≥2 guides per gene. | Rule off-target activation; phenotype should be consistent across ≥2 guides per gene. | Using 3-4 independent sgRNAs targeting different exons or promoter regions. |
II. Critical Reagents and Validation Tools
The selection of high-quality, validated reagents is fundamental. The table below catalogs essential solutions.
Table 2: The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Function & Importance | Specific Considerations for CRISPRko/i/a |
|---|---|---|
| Validated Guide RNA Libraries | Ensures comprehensive, specific, and uniform coverage of target genes or regulatory elements. | CRISPRko: Focus on early exons. CRISPRi: Target -200 to +50 bp from TSS. CRISPRa: Target -200 to +400 bp from TSS. Must be sequence-verified. |
| High-Efficiency Delivery Systems | Enables high-coverage perturbations with minimal bias. Critical for pooled screens. | Lentiviral vectors are standard. Titration to achieve low MOI (~0.3-0.5) for pooled screens is essential to avoid multiple integrations. |
| Cas9/dCas9 Effector Cell Lines | Provides stable, uniform expression of the CRISPR nuclease or effector domain. | Use clones with consistent, moderate expression. For CRISPRi/a, inducible dCas9 systems (e.g., Tet-On) allow temporal control. |
| PCR & NGS Reagents for Amplification | Enables amplification of guide barcodes or target loci for deep sequencing. | Use high-fidelity polymerases. Include unique molecular identifiers (UMIs) to control for PCR bias in screen deconvolution. |
| Antibodies for Validation | Confirm protein knockout (CRISPRko) or assess changes in histone modifications (CRISPRi/a). | For CRISPRko: Target protein antibody. For CRISPRi: H3K9me3 at target locus (ChIP). For CRISPRa: H3K27ac or RNA Pol II at target (ChIP). |
| Cell Viability & Selection Agents | Enriches for successfully transduced cells. | Puromycin is common for lentiviral selection. Concentration must be pre-titrated for each cell line. |
III. Detailed Methodologies for Key Validation Experiments
Protocol 1: Validation of Perturbation Efficiency via qRT-PCR (for CRISPRi/a)
- Cell Preparation: Generate polyclonal or monoclonal populations transduced with the target guide RNA and dCas9-effector. Include a non-targeting guide control.
- RNA Isolation: At 72-96 hours post-transduction/induction, harvest cells and isolate total RNA using a column-based kit with on-column DNase I treatment.
- cDNA Synthesis: Use 500 ng - 1 µg of total RNA with a reverse transcription kit using random hexamers.
- qPCR: Perform quantitative PCR in triplicate using SYBR Green or TaqMan assays. Design primers spanning an exon-exon junction to avoid genomic DNA amplification.
- Analysis: Calculate ∆∆Ct relative to the non-targeting guide control and a stable reference gene (e.g., GAPDH, ACTB). Expect >70% repression for CRISPRi and 5-50 fold activation for CRISPRa on competent targets.
Protocol 2: T7 Endonuclease I (T7E1) Assay for CRISPRko Indel Detection
- Genomic DNA Extraction: Isolate gDNA from edited and control cells 72-96 hours post-transduction using a silica-membrane column.
- PCR Amplification: Amplify a 300-500 bp region surrounding the guide RNA target site using a high-fidelity polymerase.
- DNA Heteroduplex Formation: Purify PCR products. Denature and reanneal in a thermocycler: 95°C for 5 min, ramp down to 85°C at -2°C/s, then to 25°C at -0.1°C/s.
- T7E1 Digestion: Incubate 200-400 ng of reannealed PCR product with 5-10 units of T7 Endonuclease I in supplied buffer for 30-60 minutes at 37°C.
- Analysis: Run digested products on a 2-2.5% agarose gel. Cleaved bands indicate the presence of indel mutations. Efficiency can be estimated by band intensity.
IV. Visualizing Experimental Workflows and Relationships
Diagram 1: Generalized CRISPR Screen & Validation Workflow
Diagram 2: Mechanism of Action: CRISPRko vs CRISPRi vs CRISPRa
CRISPRko vs CRISPRi vs CRISPRa: A Head-to-Head Comparative Analysis
Within the expanding CRISPR toolbox for functional genomics and therapeutic development, CRISPRko (knockout), CRISPRi (interference), and CRISPRa (activation) represent three principal modalities for modulating gene expression. This technical guide provides a comparative analysis of these technologies, focusing on the critical parameters of permanence, reversibility, kinetics, and magnitude of effect. The assessment is grounded in their mechanisms, experimental performance, and suitability for various research and drug development applications.
CRISPRko, CRISPRi, and CRISPRa exploit the programmable targeting of the Cas9 nuclease (or its derivatives) to a specific genomic locus but differ fundamentally in their effector domains and subsequent molecular events.
- CRISPRko (Knockout): Utilizes wild-type Streptococcus pyogenes Cas9 (spCas9) or other nucleases to create a targeted double-strand break (DSB) in the coding region of a gene. Repair via error-prone non-homologous end joining (NHEJ) leads to insertion/deletion (indel) mutations, resulting in frameshifts and premature stop codons, thereby permanently disrupting the gene.
- CRISPRi (Interference): Employs a catalytically "dead" Cas9 (dCas9) fused to transcriptional repressor domains (e.g., KRAB, SID4x). The dCas9-KRAB complex binds to the promoter or early coding region of a target gene, recruiting chromatin modifiers that establish a repressive heterochromatin state, leading to reversible transcriptional repression.
- CRISPRa (Activation): Uses dCas9 fused to transcriptional activator domains. Common systems include dCas9-VPR (a tripartite activator: VP64, p65, Rta) or dCas9-p300 (a histone acetyltransferase). These complexes recruit co-activators and modify chromatin to an open, active state, driving targeted gene upregulation. This effect is typically reversible.
Comparative Matrix: Core Parameters
The following tables synthesize quantitative and qualitative data from recent literature on the performance characteristics of these three modalities.
Table 1: Qualitative Comparison of Core Attributes
| Parameter | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Molecular Mechanism | NHEJ-mediated indel mutagenesis | dCas9-mediated transcriptional repression | dCas9-mediated transcriptional activation |
| Permanence | Permanent (genomic alteration) | Reversible (epigenetic modulation) | Reversible (epigenetic modulation) |
| Typical Onset Kinetics | Fast (DSB repair in hours; protein depletion depends on turnover) | Moderate (chromatin remodeling in 24-48 hrs) | Moderate to Slow (chromatin remodeling in 24-72 hrs) |
| Primary Application | Essential gene identification, loss-of-function screens, generating knockout models | Tunable knockdown, studying essential genes, reversible phenotype interrogation | Gain-of-function screens, overexpression phenotypes, cellular reprogramming |
| Key Advantage | Complete, permanent ablation of function. | Reversible, tunable, reduced off-target transcriptional effects vs. RNAi. | Targeted, endogenous gene activation without transgenesis. |
| Key Limitation | Cannot target essential genes in proliferating cells. Confounding off-target indels. | Repression is often incomplete (<90-95%). Effect depends on chromatin context. | Magnitude of activation is variable and gene-dependent (often 2-10x). |
Table 2: Quantitative Performance Metrics (Representative Data)
| Metric | CRISPRko | CRISPRi (dCas9-KRAB) | CRISPRa (dCas9-VPR) |
|---|---|---|---|
| Magnitude of Effect | ~100% gene disruption (biallelic) | 70-95% mRNA repression | 2- to 50-fold mRNA activation (median ~5-10x) |
| Time to Max Effect | 2-3 days (protein loss) | 2-3 days | 3-5 days |
| Reversal Kinetics (upon dCas9 loss) | Not reversible | Full reversal in 3-7 days | Full reversal in 5-10 days |
| Typical Screening Performance (Z'-factor) | High (>0.5) | Moderate to High | Moderate (more variable) |
| Off-Target Risk (Transcriptome-wide) | Low-frequency indels at off-target genomic sites. | Minimal transcriptional perturbation at off-target sites; possible squelching. | Minimal transcriptional perturbation at off-target sites; possible squelching. |
Experimental Protocols
Protocol for CRISPRko Loss-of-Function Screening
Objective: To perform a genome-wide negative selection screen to identify essential genes.
- Library Design: Use a pooled, genome-wide sgRNA library (e.g., Brunello, 4 sgRNAs/gene).
- Virus Production: Lentivirally package the sgRNA library in HEK293T cells to achieve low MOI (~0.3) ensuring single integration.
- Cell Transduction: Infect target cells (e.g., a cancer cell line) at a coverage of >500 cells per sgRNA. Select with puromycin for 3-5 days.
- Screen Passage: Maintain the population for ~14-21 population doublings. Harvest genomic DNA at Day 0 (reference) and Day 14/21 (endpoint).
- Amplification & Sequencing: PCR amplify integrated sgRNA sequences with indexed primers for NGS.
- Analysis: Align sequences to the reference library. Use algorithms (MAGeCK, pinAPL) to compare sgRNA depletion between timepoints and rank essential genes.
Protocol for CRISPRi/a Time-Course Reversibility Assay
Objective: To measure the kinetics of gene repression/activation and its reversal.
- Stable Line Generation: Create cell lines stably expressing dCas9-KRAB (i) or dCas9-VPR (a) via lentiviral transduction and blasticidin selection.
- sgRNA Transduction: Transduce stable lines with lentiviral sgRNAs targeting a gene of interest (GOI) and a non-targeting control (NTC). Use a fluorescent marker (e.g., GFP) for sorting.
- Induction/Withdrawal: For kinetics: Harvest cells at 0, 24, 48, 72, 96h post-transduction. For reversibility: At 96h, use FACS to isolate GFP+ (sgRNA-expressing) cells. Split population; maintain one with continued expression, and in the other, use doxycycline withdrawal (if using inducible dCas9) or sort for GFP- decay population.
- Readout: Quantify mRNA levels of GOI via RT-qPCR at each time point, normalized to NTC and housekeeping genes.
- Modeling: Fit curves to determine time to half-maximal effect and reversal half-life.
The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Research Reagents
| Reagent | Function in CRISPRko/i/a | Example/Supplier Notes |
|---|---|---|
| High-Efficiency Cas9/dCas9 Expression Vector | Drives consistent, high-level expression of the nuclease or effector. | lentiCas9-Blast (Addgene #52962), lenti dCas9-KRAB-Blast (Addgene #89567), lenti dCas9-VPR-Blast (Addgene #89789). |
| Validated sgRNA Cloning Backbone | Allows for efficient sgRNA cloning and expression, often with a selection marker. | lentiGuide-Puro (Addgene #52963), pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro. |
| Validated sgRNA Library | Pre-designed, pooled sets of sgRNAs for genome-wide or pathway-specific screens. | Brunello (ko), Dolcini (i), Calabrese (a) libraries from Broad Institute. |
| Lentiviral Packaging Plasmids | For production of replication-incompetent lentivirus to deliver CRISPR components. | psPAX2 (packaging) and pMD2.G (VSV-G envelope) from Addgene. |
| Transfection/Gene Delivery Reagent | For plasmid delivery into packaging or target cells. | PEI MAX, Lipofectamine 3000, Fugene HD. |
| Selection Antibiotics | To select for cells stably expressing CRISPR components. | Puromycin, Blasticidin, Hygromycin B. Quality varies; use cell-titrated concentrations. |
| NGS Library Prep Kit | For preparing sgRNA amplicons from genomic DNA for deep sequencing. | NEBNext Ultra II DNA Library Prep Kit. Index primers specific to sgRNA library are required. |
| qPCR Master Mix & Primers | For quantifying gene expression changes in CRISPRi/a experiments and validating knockouts. | SYBR Green or TaqMan assays. Design primers spanning the CRISPR target site to detect indels (for ko). |
Within the rapidly evolving field of functional genomics, the comparison of CRISPR-based knockout (CRISPRko), interference (CRISPRi), and activation (CRISPRa) technologies forms a critical research thesis. This guide provides an in-depth technical analysis of these core technologies, focusing on their precision, versatility, and practicality for researchers and drug development professionals. CRISPRko utilizes Cas9 or Cas12 nucleases to create double-strand breaks, resulting in frameshift mutations and gene disruption. CRISPRi employs a catalytically dead Cas9 (dCas9) fused to transcriptional repressors (e.g., KRAB) to epigenetically silence gene expression. CRISPRa uses dCas9 fused to transcriptional activators (e.g., VPR, SAM complex) to upregulate gene expression.
Quantitative Comparison of Core Technologies
Table 1: Performance Metrics for CRISPRko, CRISPRi, and CRISPRa
| Metric | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Knockdown Efficiency (%) | >90% (protein null) | 70-95% (mRNA reduction) | 5-50x induction (vs. baseline) |
| On-Target Specificity (Off-Target Rate) | Moderate (guide-dependent, ~0.1-60% indels at known off-targets) | High (epigenetic, minimal sequence alteration) | High (epigenetic, minimal sequence alteration) |
| Temporal Control | Irreversible | Reversible (upon degron or Dox control) | Reversible (upon degron or Dox control) |
| Multiplexing Capacity | High (up to 10+ genes with arrayed gRNAs) | Very High (dCas9 can be targeted to many loci) | Very High (dCas9 can be targeted to many loci) |
| Typical Delivery Method | Lentivirus, RNP, AAV | Lentivirus (stable dCas9 line) | Lentivirus (stable dCas9 line) |
| Key Application | Essential gene identification, loss-of-function screens | Tuning gene dosage, studying essential genes | Gain-of-function, overexpression screens |
Table 2: Strengths & Limitations: Precision, Versatility, Practicality
| Aspect | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Precision | Strength: Permanent, complete ablation. Limitation: Potential for off-target indels; genomic scarring. | Strength: Reversible, tunable, minimal off-target transcriptional effects. Limitation: Incomplete knockdown; potential for epigenetic drift. | Strength: Reversible, tunable activation. Limitation: Heterogeneous activation levels; potential for super-physiological effects. |
| Versatility | Strength: Works in dividing/non-dividing cells; vast validated gRNA libraries. Limitation: Not suitable for studying essential genes in viability screens. | Strength: Enables study of essential genes; fine-tuning of expression; facilitates temporal studies. Limitation: Requires stable dCas9-repressor cell line; efficacy depends on chromatin state. | Strength: Enables gain-of-function in native genomic context; multi-gene activation. Limitation: Requires stable dCas9-activator line; size of effector may limit delivery. |
| Practicality | Strength: Simple, robust, cost-effective; standardized protocols. Limitation: Mixed phenotypes from indels; clonal variation. | Strength: Enables genome-wide silencing screens with minimal toxicity. Limitation: More complex reagent generation; potential for dCas9 toxicity or background. | Strength: Powerful for identifying drug targets and resistance genes. Limitation: High background noise; complex vector design. |
Detailed Experimental Protocols
Protocol for a CRISPRko Pooled Screening Workflow
Aim: To conduct a genome-wide loss-of-function screen to identify genes essential for cell proliferation.
- Library Selection: Choose a validated genome-wide CRISPRko library (e.g., Brunello, ~77k gRNAs).
- Lentivirus Production: Generate lentiviral particles in HEK293T cells by co-transfecting the library plasmid with psPAX2 and pMD2.G packaging plasmids using PEI transfection reagent.
- Cell Transduction: Infect target cells at a low MOI (~0.3) to ensure single gRNA integration. Include a non-targeting control (NTC) gRNA population.
- Puromycin Selection: 48 hours post-transduction, select transduced cells with puromycin (dose determined by kill curve) for 5-7 days.
- Screen Passaging: Maintain the library-covered cell population for at least 14 population doublings. Passage cells regularly, maintaining >500x library coverage at each step.
- Genomic DNA (gDNA) Extraction: Harvest cells at the start (T0) and end (T14) of the screen. Extract gDNA using a large-scale kit (e.g., Qiagen Maxi Prep).
- gRNA Amplification & Sequencing: Perform a two-step PCR to amplify the integrated gRNA cassette from gDNA and add Illumina sequencing adapters/indexes. Pool and purify PCR products.
- Next-Generation Sequencing (NGS): Sequence on an Illumina HiSeq/NovaSeq platform to obtain >500 reads per gRNA.
- Data Analysis: Align reads to the library reference. Use MAGeCK or similar algorithms to compare gRNA abundance between T0 and T14, identifying significantly depleted gRNAs/genes.
Protocol for CRISPRi/a Knockdown/Activation Validation
Aim: To validate hits from a CRISPRi/a screen via individual gene targeting.
- Cloning: Clone individual gRNAs targeting the gene of interest (or NTC) into a sgRNA expression vector compatible with your stable dCas9-effector cell line.
- Stable Cell Line Generation (if not existing): Create a cell line stably expressing dCas9-KRAB (for i) or dCas9-VPR (for a) via lentiviral transduction and blasticidin/antibiotic selection.
- Transfection/Transduction: Deliver individual gRNA constructs into the stable dCas9-effector cell line via lipofection or lentiviral transduction.
- Efficacy Validation (qRT-PCR): 72-96 hours post-gRNA delivery, extract total RNA, synthesize cDNA, and perform qPCR with TaqMan or SYBR Green assays for the target gene. Normalize to housekeeping genes (e.g., GAPDH, ACTB).
- Phenotypic Assay: In parallel, run the relevant phenotypic assay (e.g., CellTiter-Glo for viability, flow cytometry for a marker).
Visualizations
CRISPR Pooled Screen Workflow
Core Mechanisms of CRISPRko, i, and a
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for CRISPR Functional Genomics
| Reagent Category | Specific Item/Kit | Function & Explanation |
|---|---|---|
| CRISPR Nuclease/Effector | S. pyogenes Cas9 (WT for KO), dCas9-KRAB (for i), dCas9-VPR (for a) | The core enzyme: Cas9 creates DSBs; dCas9 fusion proteins enable targeted repression or activation without DNA cutting. |
| gRNA Library & Cloning | Brunello/GeCKO v2 (KO), Dolcetto/Calabrese (i), SAM/CRISPRa libraries (a) | Pre-designed, sequence-validated pooled libraries for genome-wide screening. Cloning kits (e.g., Lentiguide, lentiArray) for arrayed validation. |
| Lentiviral Packaging | psPAX2 (packaging), pMD2.G (VSV-G envelope) plasmids | Essential 2nd/3rd generation system components to produce replication-incompetent lentiviral particles for efficient delivery. |
| Delivery & Selection | Polybrene/Hexadimethrine bromide, Puromycin, Blasticidin S | Polybrene enhances viral transduction. Antibiotics select for cells successfully expressing the delivered resistance gene. |
| Genomic DNA Extraction | Qiagen Blood & Cell Culture DNA Maxi Kit | For high-quality, high-quantity gDNA extraction from millions of screen cells, required for subsequent gRNA amplification. |
| gRNA Amplification | KAPA HiFi HotStart ReadyMix, Custom P5/P7 PCR Primers | High-fidelity polymerase ensures accurate amplification of the integrated gRNA sequence from gDNA for NGS library prep. |
| NGS & Analysis | Illumina NextSeq 2000, MAGeCK (0.5.9+), BAGEL2 | Sequencing platform and specialized computational tools for quantifying gRNA abundance and identifying significant hits. |
| Validation Assays | TaqMan Gene Expression Assays, CellTiter-Glo Luminescent Viability Assay | Gold-standard qPCR for knockdown/activation validation. Robust ATP-based assay for quantifying cell viability/proliferation. |
Within the comparative study of CRISPR knockout (CRISPRko), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa), a critical axis of analysis is their differential impact on cellular phenotype. This guide details the technical considerations for assessing cytotoxicity, fitness defects, and compensatory adaptation, which are paramount for interpreting functional genomics screens and therapeutic target validation.
The choice of perturbation modality (ko, i, a) intrinsically links to the phenotypic outcome measured. CRISPRko causes permanent DNA cleavage and frameshift mutations, leading to complete loss-of-function. CRISPRi, typically using a deactivated Cas9 (dCas9) fused to a repressive domain like KRAB, results in reversible transcriptional repression. CRISPRa, using dCas9 fused to transcriptional activators (e.g., VPR, SAM), induces gene expression. Each method presents distinct kinetic profiles, reversibility, and off-target effects, which directly influence cytotoxicity, fitness, and the propensity for genetic compensation.
Quantitative Comparison of Core Phenotypic Impacts
Table 1: Phenotypic Impact of CRISPR Modalities
| Parameter | CRISPRko | CRISPRi (dCas9-KRAB) | CRISPRa (dCas9-VPR) |
|---|---|---|---|
| Genetic Outcome | Indels, frameshifts, gene disruption | Epigenetic repression, reduced mRNA | Transcriptional activation, increased mRNA |
| Reversibility | Essentially irreversible | Reversible upon sgRNA/dCas9 removal | Reversible upon sgRNA/dCas9 removal |
| Onset Kinetics (Protein Depletion) | Days (depends on protein stability) | Hours to days | Hours to days |
| Cytotoxicity Potential | High for essential genes; can trigger p53 response | Moderate; fewer DNA damage concerns | Variable; can cause overexpression toxicity |
| Fitness Impact Measurement | Strong, definitive fitness defects | Attenuated fitness defects | Negative fitness from overexpression |
| Compensation Activation | High (genetic compensation network activation) | Lower (phenotypic buffering) | Context-dependent |
| Common Off-Target Effects | DNA double-strand breaks at off-target sites | Transcriptional repression at off-target sites | Transcriptional activation at off-target sites |
Table 2: Typical Screening Readouts from Pooled Libraries (Recent Data)
| Assay | CRISPRko (Avg. Essential Gene z-score) | CRISPRi (Avg. Essential Gene z-score) | CRISPRa (Avg. Selective Gene z-score) |
|---|---|---|---|
| Cell Viability (Proliferation) | -4.5 to -6.0 | -2.0 to -3.5 | +1.5 to +3.0 (for growth promoters) |
| Apoptosis Induction (Caspase 3/7) | High (>5-fold increase) | Moderate (2-4 fold increase) | Low (context-dependent) |
| Senescence Induction (β-galactosidase) | Variable | Low | Can be high for certain oncogenes |
| Migration/Invasion Defect | Pronounced | Partial inhibition | Often enhanced |
Experimental Protocols for Phenotypic Assessment
Protocol 1: Long-Term Competitive Fitness Assay
Purpose: Quantify the fitness defect or advantage of a perturbation over multiple cell divisions. Method:
- Infection & Selection: Transduce a pooled CRISPR library (ko, i, or a) into target cells at low MOI (<0.3) to ensure single integrations. Select with puromycin for 72-96 hours.
- Harvest Timepoints: Harvest cells for genomic DNA (gDNA) extraction at T0 (post-selection) and at regular intervals (e.g., T7, T14, T21 days). Maintain cells at sufficient coverage (500x library representation).
- gDNA Extraction & Amplification: Extract gDNA (e.g., Qiagen Blood & Cell Culture DNA Maxi Kit). Amplify integrated sgRNA sequences via a two-step PCR using barcoded primers for multiplexed NGS.
- Sequencing & Analysis: Sequence on an Illumina platform. Count sgRNA reads. Calculate fold-change and log2 fold-change for each sgRNA between Tn and T0. Generate gene-level scores (e.g., MAGeCK, CERES for CRISPRko; drugZ for CRISPRi/a). Key Reagents: Pooled sgRNA library, lentiviral packaging plasmids, polybrene, puromycin, gDNA extraction kit, Q5 High-Fidelity DNA Polymerase, Illumina sequencing platform.
Protocol 2: Acute Cytotoxicity & Apoptosis Measurement
Purpose: Distinguish rapid cytotoxic events from chronic fitness defects. Method:
- Arrayed Perturbation: Seed cells in 96-well plates. Transfert with individual sgRNAs + Cas9/dCas9 constructs or transduce with pre-packaged lentivirus for single perturbations.
- Viability Readouts: At 72-96 hours post-perturbation, assay using:
- ATP-based Luminescence: Add CellTiter-Glo reagent, measure luminescence.
- Membrane Integrity: Perform LDH release assay (CytoTox-96).
- Apoptosis-Specific Readout: At 48-72 hours, use a Caspase-3/7 Glo assay or stain with Annexin V-FITC/PI for flow cytometry analysis. Key Reagents: Arrayed sgRNAs, lipofectamine or similar, CellTiter-Glo, LDH assay kit, Caspase-3/7 Glo, Annexin V binding buffer.
Protocol 3: Monitoring Genetic Compensation (CRISPRko-specific)
Purpose: Identify transcriptional upregulation of homologous genes following knockout. Method:
- Generate Knockout Clones: Create isogenic knockout and control cell lines using CRISPRko.
- RNA-Seq Analysis: Perform total RNA sequencing (RNA-seq) in triplicate for knockout and control lines. Use a depth of 30-50 million reads per sample.
- Bioinformatic Pipeline: Align reads (STAR, HISAT2), quantify gene expression (featureCounts), perform differential expression analysis (DESeq2). Focus on significant upregulation (FDR < 0.05, log2FC > 1) of paralogs or genes in related pathways.
- Functional Rescue Validation: Use CRISPRi to knockdown the upregulated compensatory gene(s) in the knockout background and reassess the original phenotypic defect. Key Reagents: RNA extraction kit (e.g., RNeasy), RNA-seq library prep kit, DESeq2 software package.
Visualizing Mechanisms and Workflows
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Phenotypic CRISPR Screening
| Reagent / Solution | Function & Rationale | Example Product (Reference) |
|---|---|---|
| Arrayed or Pooled sgRNA Libraries | Target-specific guide RNA sequences for ko, i, or a. Library design is critical for on-target efficiency and minimizing off-targets. | Synthego Arrayed Libraries, Brunello CRISPRko Library, Calabrese CRISPRi/a Libraries |
| dCas9 Effector Plasmids | Express the nuclease-deficient Cas9 fused to transcriptional modulators. | pLV hU6-sgRNA hUbC-dCas9-KRAB (Addgene #71236) for CRISPRi; dCas9-VPR for CRISPRa. |
| High-Efficiency Transfection/Transduction Reagents | For delivery of RNP, plasmid, or viral particles into target cells. Cell-type specific optimization required. | Lipofectamine CRISPRMAX (RNP), Polybrene (lentiviral transduction), FuGENE HD. |
| Next-Generation Sequencing Kits | For amplicon sequencing of sgRNA barcodes from pooled screens. | Illumina Nextera XT, NEBNext Ultra II DNA Library Prep. |
| Cell Viability & Cytotoxicity Assays | Quantitatively measure phenotypic outcomes like proliferation, death, and metabolic activity. | CellTiter-Glo 3D (3D cultures), RealTime-Glo MT Cell Viability Assay (kinetics), LDH-Glo Cytotoxicity Assay. |
| gDNA Extraction Kits (Large Scale) | High-yield, high-quality genomic DNA extraction from millions of cells for pooled screen representation. | Qiagen Blood & Cell Culture DNA Midi/Maxi Kit. |
| Bioinformatics Analysis Pipelines | Software to process NGS data, calculate sgRNA depletion/enrichment, and assign gene-level phenotypic scores. | MAGeCK-VISPR, CERES (for CRISPRko correction), PinAPL-Py. |
In the functional genomics revolution driven by CRISPR-based technologies, the critical challenge is the accurate interpretation of phenotypic data. Distinguishing between direct, on-target consequences of gene perturbation and secondary, indirect cellular adaptations is paramount for validating therapeutic targets. This guide provides a technical framework for this discrimination, contextualized within the comparative use of CRISPR knockout (CRISPRko), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa).
1. Mechanistic Foundations and Data Interpretation Challenges
Each CRISPR modality induces distinct temporal and mechanistic effects, inherently influencing the prevalence of direct vs. indirect outcomes.
| Perturbation Modality | Molecular Effect | Key Interpretational Challenge |
|---|---|---|
| CRISPRko | Permanent DNA cleavage, frameshift mutations, and functional gene deletion. | Phenotypes can be confounded by compensatory adaptations or clonal selection over time. |
| CRISPRi | Reversible, transcriptional repression via dCas9-KRAB fusion. | Potential for incomplete silencing; phenotypes may reflect partial loss-of-function. |
| CRISPRa | Targeted transcriptional upregulation via dCas9-VPR fusion. | Overexpression artifacts, neo-/misexpression, and feedback inhibition can obscure direct effects. |
2. Experimental Protocols for Disambiguation
A multi-pronged experimental strategy is required to isolate direct effects.
Protocol 2.1: Temporal Phenotyping & Kinetic Analysis
- Objective: Differentiate immediate primary effects from slower adaptive responses.
- Methodology: Utilize a doxycycline-inducible dCas9 system (for CRISPRi/a) or a Cre-inducible Cas9 system (for CRISPRko). Post-induction, collect multi-omics data (e.g., RNA-seq, phospho-proteomics) at dense time points (e.g., 6h, 12h, 24h, 48h, 72h, 7d). Direct transcriptional targets will show significant changes early (e.g., by 24h), while indirect feedback loops manifest later.
- Key Control: Non-targeting sgRNA in the same inducible system.
Protocol 2.2: Orthogonal Validation with Acute Protein Degradation
- Objective: Corroborate findings with a mechanistically distinct perturbation.
- Methodology: For a hit from a CRISPRko screen, engineer an orthogonal degron system (e.g., auxin-inducible degron or dTAG) at the endogenous locus of the protein of interest. Perform acute protein depletion (over 2-24 hours) and measure the same phenotypic endpoint (e.g., viability, reporter activity). A concordant phenotype strongly suggests a direct effect.
- Key Control: Parental cell line lacking the degron system, treated identically.
Protocol 2.3: Multi-Modal Perturbation Concordance
- Objective: Filter for robust, modality-independent phenotypes.
- Methodology: Target the same genomic locus (e.g., gene promoter) with CRISPRko, CRISPRi, and CRISPRa in parallel experiments. Assay the phenotype (e.g., cell proliferation, differentiation marker). Direct effects are indicated by complementary outcomes: growth inhibition with ko/i and enhancement with a for an essential oncogene.
- Key Control: Non-targeting sgRNAs for each dCas9 fusion variant.
3. The Scientist's Toolkit: Essential Research Reagents
| Reagent / Solution | Function in Disambiguation Experiments |
|---|---|
| Inducible dCas9 Cell Line (Tet-On) | Enables kinetic studies by allowing precise temporal control over CRISPRi/a perturbation. |
| CRISPRko, i, a sgRNA Library (Targeting Same Gene Set) | Allows parallel screening to identify modality-concordant phenotypes, filtering for direct effects. |
| Auxin-Inducible Degron (AID) Tagging System | Provides orthogonal, acute protein depletion for rapid validation of genetic hits. |
| Single-Cell Multi-omic Sequencing Platform (CITE-seq, Perturb-seq) | Maps genotype-to-phenotype relationships in pooled screens, revealing indirect network states. |
| dCas9 Fusion Variants (KRAB, VPR, DNMT3A) | Mechanistically distinct epigenetic modulators to test phenotype consistency across perturbation types. |
4. Data Presentation & Quantitative Comparison
The following table summarizes hypothetical quantitative outcomes from a multi-modal screen targeting a putative tumor suppressor gene (TSG1) and a metabolic housekeeping gene (HK1), illustrating data patterns that signal direct vs. indirect effects.
| Target Gene | Perturbation | Phenotype: Cell Growth (Log2 Fold Change) | Phenotype: Key Pathway Marker (Protein Level) | Interpretation |
|---|---|---|---|---|
| TSG1 | CRISPRko | +0.85 | p-ERK: Increased | Direct: Consistent gain-of-function across modalities. |
| CRISPRi | +0.72 | p-ERK: Increased | ||
| CRISPRa | -0.65 | p-ERK: Decreased | ||
| HK1 | CRISPRko | -1.50 (at 7d) | ROS: High | Indirect: Discordant results; ko suggests essentiality not supported by i, indicating potential adaptive lethality. |
| CRISPRi | -0.15 | ROS: Moderate | ||
| CRISPRa | +0.10 | ROS: Low | ||
| Indirect Mediator X | CRISPRko | -0.95 | p53: Stabilized | Validated Indirect: Phenotype only appears in late ko, not acute i. Correlates with secondary adaptation. |
| CRISPRi (Acute) | +0.05 | p53: No change |
5. Visualizing Logical & Pathway Relationships
Temporal Logic of Direct vs. Indirect Effects
Core Mechanistic Differences: CRISPRko vs i vs a
This whitepaper provides a technical framework for selecting the optimal CRISPR-based perturbation modality—CRISPRko (knockout), CRISPRi (interference), or CRISPRa (activation)—based on the fundamental biological question of loss-of-function (LOF) vs. gain-of-function (GOF) analysis. The choice is not merely technical but foundational, dictating the physiological relevance, interpretability, and success of functional genomics and drug target validation studies.
Core Mechanistic & Quantitative Comparison
The three modalities enable distinct genetic perturbations by leveraging catalytically altered Cas9 fused to different effector domains.
Table 1: Quantitative & Functional Comparison of CRISPRko, CRISPRi, and CRISPRa
| Parameter | CRISPRko (LOF) | CRISPRi (LOF) | CRISPRa (GOF) |
|---|---|---|---|
| Core Mechanism | Nuclease-active Cas9 (e.g., SpCas9) induces double-strand breaks, leading to frameshift indels and gene knockout. | Catalytically dead Cas9 (dCas9) fused to transcriptional repressors (e.g., KRAB) binds near transcription start site (TSS) to block initiation or elongation. | dCas9 fused to transcriptional activators (e.g., VPR, SAM) binds to promoter/enhancer regions to recruit RNA Pol II and increase transcription. |
| Efficiency (Typical Range) | >80% protein knockout in polyclonal populations. | 70-95% transcript knockdown, varies by target and guide design. | 2- to 50-fold transcript upregulation; highly target-dependent. |
| Key Effect | Permanent, complete genetic ablation. | Reversible, tunable (via inducer dosage), partial transcript knockdown. | Inducible, tunable transcript overexpression. |
| Primary Use Case | Essential gene identification, non-essential gene validation, synthetic lethality screens. | Titratable knockdown, study of essential genes where knockout is lethal, study of multi-gene families with redundancy. | Gene overexpression screens, suppressor screens, modeling oncogenic GOF, endogenous gene activation for therapeutic mimicry. |
| Major Artifact Concerns | Confounding DNA damage response (p53 activation), clonal selection bias, genomic rearrangements. | Off-target transcriptional repression, "squelching" due to high KRAB expression, potential epigenetic memory. | Off-target gene activation, super-enhancer interference, toxicity from extreme overexpression. |
| Typical Screening Readiness | 7-14 days post-transduction for negative selection screens; robust for dropout phenotypes. | 5-10 days; suitable for acute and temporal knockdown studies. | 3-7 days for positive selection screens; requires optimization of activator strength. |
Decision Framework: LOF vs. GOF Biological Questions
LOF Questions (Use CRISPRko or CRISPRi):
- Gene Essentiality: Which genes are required for cell proliferation/survival under a specific condition?
- Pathway Necessity: Is a specific gene/pathway necessary for a phenotypic outcome (e.g., drug response, differentiation)?
- Functional Redundancy: Does silencing (rather than ablating) a gene family member reveal a phenotype?
GOF Questions (Use CRISPRa):
- Gene Sufficiency: Can overexpression of a gene drive a phenotypic change (e.g., resistance, differentiation, proliferation)?
- Suppressor Identification: Which genes, when overexpressed, rescue a disease phenotype or drug toxicity?
- Enhancer/Activator Discovery: Which non-coding genomic elements, when activated, influence target gene expression?
Selection Protocol:
- Define Phenotype: Is the expected hit a dropout (LOF) or a enrichment (GOF)?
- Assess Reversibility/Tunability: Is a graded, reversible response (CRISPRi) needed, or is a binary, permanent knockout (CRISPRko) acceptable?
- Consider Gene Context: For essential genes, use CRISPRi for titratable knockdown. For genes with haploinsufficiency, CRISPRi may be preferable to CRISPRko.
- Validate with Orthogonal Methods: Hit genes from CRISPRi/a screens should be confirmed with CRISPRko (for LOF) or cDNA overexpression (for GOF).
Experimental Protocols for Key Applications
Protocol 1: Genome-Wide LOF Dropout Screen (CRISPRko)
- Library: Use Brunello or similar genome-wide sgRNA library (~4-6 guides/gene, ~75,000 guides total).
- Transduction: Lentivirally transduce target cells at an MOI of ~0.3 to ensure most cells receive 1 guide. Maintain at 500x library coverage.
- Selection & Passaging: Apply puromycin selection (1-3 μg/mL, 3-7 days). Split cells into control and experimental arms (e.g., + drug). Passage cells, maintaining 500x coverage, for 14-21 population doublings.
- Genomic DNA Extraction & NGS: Harvest cells at T0 and Tfinal. Extract gDNA, amplify sgRNA region via PCR, and sequence on Illumina platform.
- Analysis: Use MAGeCK or CRISPResso2 to calculate guide depletion/enrichment and identify essential genes.
Protocol 2: Targeted GOF Positive Selection Screen (CRISPRa)
- Library: Use a targeted CRISPRa library (e.g., Calabrese, SAM) focusing on specific gene sets (e.g., kinase, epigenetic regulators).
- Cell Line Engineering: Stable cell line expressing dCas9-VPR or SAM component (MS2-p65-HSF1) is required prior to library transduction.
- Transduction & Selection: Transduce at low MOI (<0.3). Select with appropriate antibiotics.
- Phenotype Application: Apply selective pressure (e.g., low-dose drug, nutrient stress) 5-7 days post-transduction.
- Harvest & Analysis: Harvest surviving cell population after 2-3 weeks. Process for NGS as in Protocol 1. Analyze for significantly enriched sgRNAs.
Visualizations
Title: Decision Flow: Selecting CRISPR Perturbation Modality
Title: Core Mechanisms of CRISPRko, CRISPRi, and CRISPRa
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for CRISPR Perturbation Studies
| Reagent / Material | Function & Description | Example/Supplier |
|---|---|---|
| Lentiviral sgRNA Libraries | Pre-cloned, pooled sgRNA constructs for genome-wide or targeted screens. Essential for screening scale experiments. | Addgene: Brunello (KO), Dolcetto (i), Calabrese (a). |
| dCas9 Fusion Constructs | Engineered Cas9 variants: dCas9-KRAB for CRISPRi, dCas9-VPR for CRISPRa. Required to establish the perturbation platform. | Addgene plasmids #71236 (dCas9-KRAB), #63798 (dCas9-VPR). |
| Lentiviral Packaging Mix | Plasmids (psPAX2, pMD2.G) for producing replication-incompetent lentivirus to deliver CRISPR components. | Standard third-generation packaging system. |
| Polybrene (Hexadimethrine Bromide) | Cationic polymer that enhances viral transduction efficiency by neutralizing charge repulsion. | Typically used at 4-8 μg/mL during transduction. |
| Puromycin / Selection Antibiotics | Selective agents for enriching successfully transduced cells expressing the sgRNA/CRISPR construct. | Concentration must be titrated for each cell line. |
| NGS Library Prep Kit (for sgRNA) | Optimized kits for amplifying and preparing the integrated sgRNA region from genomic DNA for sequencing. | Illumina CRISPR sgRNA Library Amplicon Kit. |
| Validated Control sgRNAs/Plasmids | Positive (essential gene) and negative (non-targeting) control guides for benchmarking perturbation efficiency. | Often included in commercial libraries or available as sets. |
| Cell Line Engineering Services | For generating stable, inducible dCas9-expressing cell lines, which can be a major technical hurdle. | Many CROs offer custom cell line generation. |
Cost, Time, and Resource Considerations for Large-Scale Screens
Within functional genomics, large-scale genetic perturbation screens are indispensable for mapping gene function and identifying therapeutic targets. The choice of CRISPR technology—CRISPR knockout (CRISPRko), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa)—fundamentally impacts experimental design, outcomes, and resource allocation. This guide details the pragmatic considerations of cost, time, and resources for deploying these technologies in genome-scale screens, a critical component of the broader thesis comparing their mechanistic and phenotypic differences.
Core Technology Comparison & Quantitative Considerations
The operational differences between CRISPRko, CRISPRi, and CRISPRa stem from their mechanisms: CRISPRko creates permanent gene disruption via double-strand breaks and indels; CRISPRi uses a catalytically dead Cas9 (dCas9) fused to a repressive domain (e.g., KRAB) to transcriptionally silence genes; CRISPRa uses dCas9 fused to transcriptional activators (e.g., VPR, SAM) to upregulate gene expression.
Table 1: High-Level Comparison of CRISPR Modalities for Genome-Scale Screens
| Consideration | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Primary Mechanism | NHEJ-mediated indel formation | dCas9-KRAB blocks transcription | dCas9-activator recruits RNA Pol II |
| Genetic Outcome | Permanent loss-of-function | Reversible knockdown | Gain-of-function/overexpression |
| Typical Efficacy (Gene Knockdown/Up) | ~80-100% (protein null) | ~70-95% (mRNA reduction) | ~5-20x (mRNA induction) |
| Key Reagent Cost (Library + Viral) | Baseline | ~10-15% higher than KO (dCas9 line) | ~20-30% higher than KO (dCas9-activator line) |
| Screen Timeline (from design to seq.) | ~10-12 weeks | ~12-14 weeks (includes dCas9 line validation) | ~12-15 weeks (includes dCas9-activator line validation) |
| Optimal Screen Type | Essentiality, resistance/sensitivity | Essentiality (tunable), synthetic lethality | Suppressor, resistance (overexpression) |
| Major Technical Risk | Off-target indels, copy-number effects | Epigenetic variegation, incomplete repression | Off-target activation, high baseline noise |
| Data Analysis Complexity | Moderate | Moderate | High (requires stringent controls for background) |
Table 2: Estimated Cost Breakdown for a Genome-Scale Human Screen (~20M cells, 5x coverage)
| Cost Category | CRISPRko | CRISPRi/a (excluding stable line gen.) |
|---|---|---|
| sgRNA Library Synthesis (80,000 sgRNAs) | $4,000 - $7,000 | $4,500 - $8,000 (often more complex promoters) |
| Lentivirus Production (Large-scale) | $2,000 - $3,500 | $2,000 - $3,500 |
| Cell Culture & Transduction Reagents | $5,000 - $8,000 | $5,000 - $8,000 |
| Next-Generation Sequencing (Multiplexed) | $3,000 - $5,000 | $3,000 - $5,000 |
| Stable Cell Line Generation (dCas9) | N/A (use WT-Cas9) | $1,500 - $3,000 (additional selection/validation) |
| Total Approximate Cost | $14,000 - $23,500 | $16,000 - $27,500 |
Experimental Protocols for Core Screening Steps
3.1. Stable Cell Line Generation (For CRISPRi/a)
- Objective: Create a polyclonal cell population stably expressing dCas9-KRAB (CRISPRi) or dCas9-VPR (CRISPRa) with uniform, moderate expression.
- Protocol:
- Lentiviral Transduction: Transduce target cells (e.g., K562, HeLa) with lentivirus carrying the dCas9-effector construct (under a constitutive promoter like EF1α) and a puromycin resistance gene at a low MOI (~0.3-0.5) to ensure single integrations.
- Selection: 48 hours post-transduction, begin selection with puromycin (e.g., 1-2 µg/mL). Maintain selection for 7-10 days.
- Validation: Confirm dCas9 expression by western blot (anti-Cas9 antibody) and functional validation using a pilot screen with known essential gene sgRNAs (for CRISPRi) or a fluorescent reporter assay (for CRISPRa).
3.2. Genome-Scale Screen Workflow
- Objective: Identify genes affecting cell fitness under a selective condition.
- Protocol:
- Library Amplification & LV Production: Amplify the plasmid sgRNA library (e.g., Brunello for KO, Dolcetto for i, Calabrese for a) and produce high-titer lentivirus in HEK293T cells.
- Cell Transduction: Transduce the screening cells (Cas9+ or dCas9-effector+) at an MOI of ~0.3 to ensure most cells receive one sgRNA. Include a non-targeting control sgRNA population.
- Selection & Expansion: 48h post-transduction, select with puromycin for 5-7 days to remove untransduced cells. Passage cells, maintaining a representation of >500 cells per sgRNA.
- Phenotype Application: Split cells into experimental (e.g., drug treatment) and control (DMSO) arms. Passage for 14-21 population doublings to allow phenotype manifestation.
- Genomic DNA Extraction & sgRNA Amplification: Harvest >50M cells per arm. Extract gDNA and PCR-amplify sgRNA regions with barcoded primers for multiplexed sequencing.
- Sequencing & Analysis: Sequence on an Illumina NextSeq. Align reads to the library reference and use MAGeCK or PinAPL-Py to calculate sgRNA depletion/enrichment and gene-level scores.
Visualizations
CRISPR Screen Experimental Workflow
Mechanisms of CRISPRko, CRISPRi, and CRISPRa
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Large-Scale CRISPR Screens
| Reagent / Material | Function & Purpose | Example Product/Note |
|---|---|---|
| Validated sgRNA Library | Pre-designed, pooled sgRNA sets targeting the genome with controls. Essential for screen integrity. | Broad Institute GPP libraries (Brunello KO, Dolcetto i, Calabrese a). |
| Lentiviral Packaging Mix | Produces high-titer, replication-incompetent lentivirus for sgRNA delivery. | psPAX2 & pMD2.G plasmids or commercial kits (e.g., Lenti-X). |
| Stable dCas9 Cell Line | For CRISPRi/a; provides uniform, inducible expression of the dCas9-effector protein. | Commercially available lines (e.g., Horizon Dharmacon) or generate in-house. |
| Puromycin Dihydrochloride | Antibiotic for selecting transduced cells expressing the sgRNA vector's resistance marker. | Critical for maintaining library representation post-transduction. |
| Polybrene (Hexadimethrine Bromide) | Enhances viral transduction efficiency by neutralizing charge repulsion. | Use at low concentration (e.g., 8 µg/mL) to avoid toxicity. |
| Next-Gen Sequencing Kit | For preparing the amplified sgRNA pool for Illumina sequencing. | NEBNext Ultra II DNA Library Prep Kit. |
| gDNA Extraction Kit (Large-Scale) | High-yield, high-quality genomic DNA extraction from >50M pelleted cells. | Qiagen Blood & Cell Culture DNA Maxi Kit. |
| Analysis Software | Computationally identifies enriched/depleted sgRNAs and scores gene significance. | MAGeCK, PinAPL-Py, or custom R/python pipelines. |
Conclusion
CRISPRko, CRISPRi, and CRISPRa are complementary pillars of modern functional genomics. CRISPRko offers definitive, permanent knockout ideal for core fitness genes. CRISPRi provides tunable, reversible knockdown with minimal off-target effects, perfect for essential gene studies and dynamic systems. CRISPRa enables gain-of-function insights, revealing gene dosage effects and activation pathways. The optimal choice depends entirely on the biological question, desired phenotype, and experimental model. Future integration with single-cell multi-omics, base editing, and in vivo delivery systems will further expand their power, solidifying their role in accelerating basic discovery and the development of novel therapeutics.