Decoding the Genome: A Comprehensive Guide to CRISPR-Cas9 Knockout Screens
This guide provides researchers, scientists, and drug development professionals with a detailed exploration of CRISPR-Cas9 knockout screen principles.
Decoding the Genome: A Comprehensive Guide to CRISPR-Cas9 Knockout Screens
Abstract
This guide provides researchers, scientists, and drug development professionals with a detailed exploration of CRISPR-Cas9 knockout screen principles. It covers the foundational biology and historical evolution of the technology, outlines current best practices for experimental design and library construction, addresses common challenges and advanced optimization strategies, and critically compares knockout screens to alternative functional genomic approaches. The article aims to be a definitive resource for planning, executing, and interpreting high-throughput genetic loss-of-function studies.
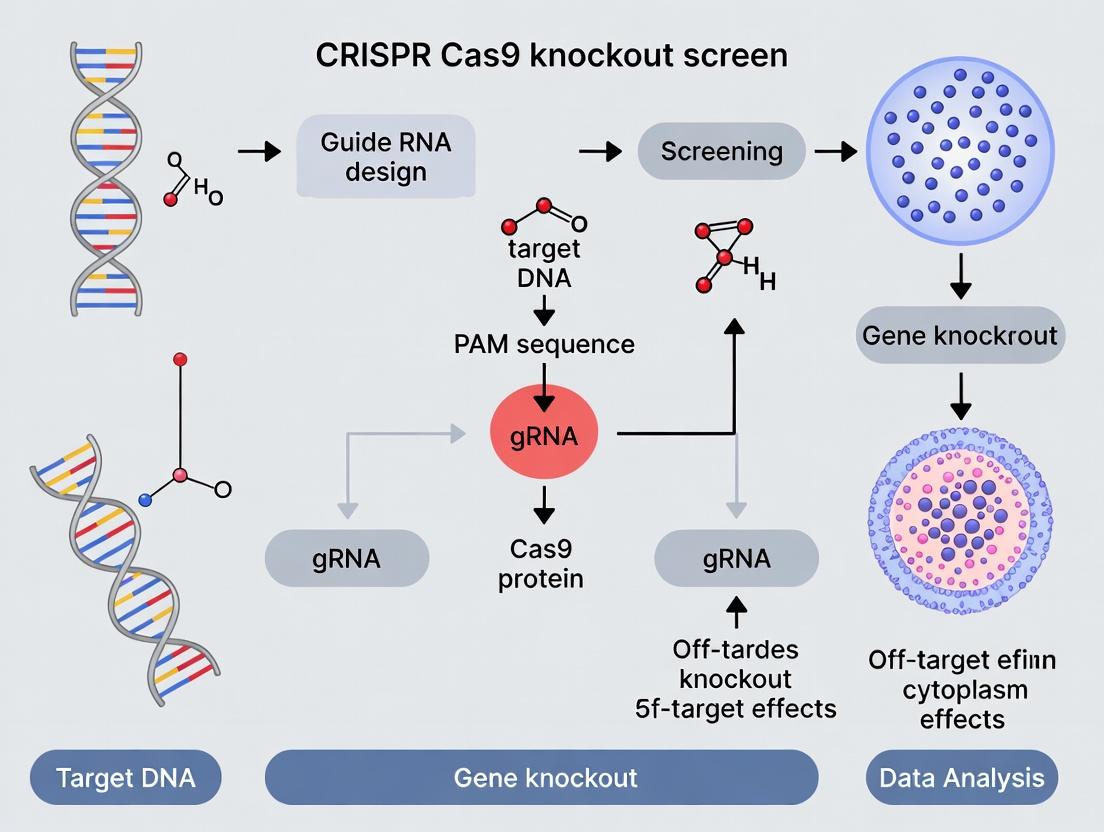
The Foundational Biology of CRISPR-Cas9 Knockout Screens: From Bacterial Immunity to Genome-Wide Discovery
Within the broader thesis on CRISPR-Cas9 knockout screen principle research, understanding the core molecular mechanism is foundational. CRISPR-Cas9-mediated gene knockout is a genome editing technique that utilizes a bacterially-derived RNA-guided endonuclease to create targeted double-strand breaks (DSBs) in genomic DNA. These breaks are predominantly repaired via the error-prone non-homologous end joining (NHEJ) pathway, leading to small insertions or deletions (indels) that can disrupt the coding sequence of a gene, resulting in a functional knockout.
Core Molecular Mechanism
System Components
The CRISPR-Cas9 system requires two core components:
- Cas9 Nuclease: The effector protein that cuts the DNA. The most commonly used variant is Streptococcus pyogenes Cas9 (SpCas9).
- Guide RNA (gRNA): A chimeric RNA molecule comprising:
- CRISPR RNA (crRNA) sequence: A 20-nucleotide spacer sequence complementary to the target DNA site.
- Trans-activating CRISPR RNA (tracrRNA) scaffold: Required for Cas9 binding and stabilization.
Target Recognition and Cleavage
The mechanism proceeds through a series of defined steps:
- Complex Formation: The gRNA binds to Cas9, forming a ribonucleoprotein (RNP) complex.
- Target Search: The RNP scans the genome for a protospacer adjacent motif (PAM). For SpCas9, the PAM sequence is 5'-NGG-3'.
- DNA Unwinding: Upon PAM recognition, Cas9 unwinds the DNA duplex.
- R-Loop Formation & Hybridization: The crRNA spacer hybridizes to the complementary DNA strand (target strand), displacing the non-complementary strand and forming an R-loop structure.
- Cleavage: Cas9 mediates a DSB ~3-4 nucleotides upstream of the PAM. The HNH nuclease domain cleaves the DNA strand complementary to the gRNA, and the RuvC-like domain cleaves the non-complementary strand.
DNA Repair and Knockout Generation
The cellular DNA repair response to the DSB determines the outcome:
- Non-Homologous End Joining (NHEJ): The dominant, error-prone pathway in most mammalian cells. NHEJ ligates the broken ends together, often resulting in small, random indels at the cleavage site. Indels that are not multiples of three cause frameshift mutations, leading to premature stop codons and gene knockout via nonsense-mediated decay (NMD) or truncation of the protein.
- Homology-Directed Repair (HDR): A precise repair pathway that uses a homologous DNA template, which can be co-delivered to introduce specific edits. In standard knockout experiments, this pathway is suppressed or not utilized.
Diagram: CRISPR-Cas9 Mechanism and Knockout Pathway
Key Quantitative Data in Knockout Screens
Table 1: Critical Parameters for Effective CRISPR Knockout Screen Design
| Parameter | Typical Range/Value | Impact on Experiment |
|---|---|---|
| gRNA Length (spacer) | 20 nucleotides | Specificity and on-target activity. |
| PAM Sequence (SpCas9) | 5'-NGG-3' | Defines genomic targeting space (~1 site per 8 bp). |
| On-Target Efficacy | 50-90% indels (varies by site) | Determines knockout efficiency in pooled population. |
| Library Size (Genome-wide) | ~70,000 - 200,000 gRNAs | Covers 3-10 gRNAs per gene; includes non-targeting controls. |
| Screen Coverage | 500-1000x cells per gRNA | Ensures statistical power and representation. |
| NHEJ Efficiency | >90% of DSB repairs | Favors knockout-inducing indels over precise HDR. |
| Indel Spectrum | -1 to -10 bp deletions most common | Frameshift probability >70% for effective knockouts. |
Table 2: Comparison of Common Cas9 Variants for Knockouts
| Cas9 Variant | PAM Sequence | Targetable Sites (Human Genome) | Key Feature for Screens |
|---|---|---|---|
| SpCas9 (Wild-type) | 5'-NGG-3' | ~9.6 million (1 in 8 bp) | Standard, well-validated. |
| SpCas9-NG | 5'-NG-3' | ~21 million (1 in 4 bp) | Expanded targeting range. |
| xCas9(3.7) | 5'-NG, GAA, GAT-3' | ~3.6 million | Broader PAM, high fidelity. |
Detailed Experimental Protocol: A Lentiviral Pooled CRISPR Knockout Screen
This protocol outlines the core workflow for a positive selection fitness screen (e.g., identifying genes essential for cell proliferation).
Materials and Reagent Preparation
- CRISPR Library: Lentiviral plasmid pool (e.g., Brunello, GeCKO v2).
- Cells: Adherent or suspension cells amenable to lentiviral transduction (e.g., HEK293T, K562).
- Lentiviral Packaging: psPAX2 (packaging) and pMD2.G (VSV-G envelope) plasmids.
- Transfection Reagent: Polyethylenimine (PEI) or commercial equivalent.
- Culture Media & Supplements: Appropriate complete medium, puromycin.
- Buffers: PBS, lysis buffer for genomic DNA extraction.
- PCR Reagents: Primers for amplifying gRNA inserts, high-fidelity polymerase.
- Sequencing: Kit for NGS library preparation, Illumina platform.
Procedure
Part A: Lentiviral Production & Titering (Days 1-4)
- Seed HEK293T cells in a 10-cm dish to reach 70-80% confluence at transfection.
- Co-transfect with the library plasmid pool, psPAX2, and pMD2.G using PEI.
- Change media 6-8 hours post-transfection.
- Harvest viral supernatant at 48 and 72 hours, filter (0.45 µm), aliquot, and store at -80°C.
- Titer Determination: Transduce target cells with serial dilutions of virus in the presence of polybrene (8 µg/mL). Select with puromycin (dose determined by kill curve) for 3-5 days. Calculate titer (TU/mL) based on percentage of surviving cells and dilution factor.
Part B: Library Transduction at Low MOI (Days 5-7)
- Seed target cells. Perform transduction at an MOI of ~0.3-0.4 to ensure most cells receive only one gRNA, with a minimum of 500 cells per gRNA in the library for coverage.
- Include polybrene (if applicable) or other transduction enhancers.
- Replace medium 24 hours post-transduction.
Part C: Selection and Cell Passaging (Days 8-20+)
- Begin puromycin selection 48-72 hours post-transduction. Maintain selection for 3-7 days until all cells in a non-transduced control are dead.
- After selection, continue to passage cells, maintaining representation (keep at least 500 cells per original gRNA at all times). For a positive selection screen, passage cells for 14-21 population doublings to allow phenotypic depletion.
Part D: Genomic DNA Extraction & gRNA Amplification (Day 21+)
- Harvest a minimum of ~1e7 cells (or equivalent genomic DNA) at the initial (T0) and final (T_f) time points. Pellet and freeze.
- Extract genomic DNA using a large-scale kit (e.g., Qiagen Maxi Prep). Ensure high yield and purity.
- Perform a two-step PCR to amplify the integrated gRNA cassette from the genomic DNA and attach Illumina sequencing adapters and sample barcodes. Use a high-fidelity polymerase to minimize bias.
- PCR1: Amplify gRNA region from genomic DNA (20-25 cycles).
- PCR2: Add full adapter sequences (10-12 cycles).
Part E: Next-Generation Sequencing & Analysis
- Purify PCR products, quantify, and pool equimolarly.
- Sequence on an Illumina platform (e.g., NextSeq, 75 bp single-end).
- Bioinformatics Analysis:
- Align reads to the library reference.
- Count gRNA reads in T0 and Tf samples.
- Use statistical packages (e.g., MAGeCK, CRISPResso2) to compare gRNA abundance between T0 and Tf, identifying significantly depleted (essential) or enriched (negative fitness) genes.
Diagram: Pooled CRISPR Knockout Screen Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for CRISPR-Cas9 Knockout Screens
| Reagent / Solution | Function & Rationale |
|---|---|
| Validated CRISPR Knockout Library (e.g., Brunello) | Pre-designed, pooled gRNA library targeting the human genome with high on-target and low off-target scores; ensures screen comprehensiveness and reproducibility. |
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Second- generation system for producing replication-incompetent, high-titer lentivirus capable of stably integrating the gRNA expression cassette into dividing and non-dividing cells. |
| Polyethylenimine (PEI), Linear, 25kDa | High-efficiency, low-cost cationic polymer transfection reagent for co-delivering library and packaging plasmids into producer cells (e.g., HEK293T) during viral production. |
| Hexadimethrine Bromide (Polybrene) | A cationic polymer that reduces charge repulsion between viral particles and cell membranes, enhancing transduction efficiency across many cell types. |
| Puromycin Dihydrochloride | Selection antibiotic. Cells expressing the lentiviral vector (with puromycin resistance gene) survive, enabling purification of successfully transduced cell populations. |
| High-Fidelity PCR Polymerase (e.g., Q5, KAPA HiFi) | Crucial for the unbiased amplification of gRNA sequences from genomic DNA during NGS library prep. Minimizes amplification errors and skewing of gRNA representation. |
| Genomic DNA Extraction Kit (Maxi/Midi Prep) | For high-yield, high-purity gDNA isolation from millions of pelleted screen cells. Purity is critical for subsequent efficient PCR amplification. |
| Illumina Sequencing Kit (e.g., NextSeq 500/550 High Output) | Provides the chemistry for clonal amplification and sequencing of the pooled gRNA amplicon library, generating millions of reads for quantitative analysis. |
This technical guide details the core principles of CRISPR-Cas9 knockout screens, focusing on the critical intersection of gRNA design and the cellular DNA repair pathways that dictate mutagenic outcomes. The efficacy of any genetic screen hinges on maximizing the probability that a targeted double-strand break (DSB) results in a complete loss-of-function allele.
Core Principles of gRNA Design
A well-designed single guide RNA (sgRNA) is the linchpin for efficient Cas9-mediated knockout. Key quantitative parameters are summarized below.
Table 1: Key Parameters for Optimal gRNA Design
| Parameter | Optimal Range/Value | Rationale & Impact on Efficiency |
|---|---|---|
| GC Content | 40-60% | Influences stability and binding affinity. Low GC (<20%) reduces efficiency; high GC (>80%) may increase off-target risk. |
| On-Target Score | >70 (tool-dependent) | Predicts cleavage efficiency. Tools use different algorithms (e.g., Doench '16, Moreno-Mateos). |
| Off-Target Score | Minimize (Max # mismatches ≥3) | Predicts specificity. Requires searching genome for sequences with ≤3 mismatches, especially in the seed region (PAM-proximal 12 bases). |
| Seed Region Sequence | No homopolymers, high specificity | Critical for R-loop stability. Mismatches here severely reduce cleavage. |
| Target Location | Early constitutive exons | Maximizes chance of frameshift leading to premature termination codon (PTC). |
| PolyT/TTTT Avoidance | Mandatory | Acts as an RNA Polymerase III termination signal in U6-driven expression systems. |
Experimental Protocol: gRNA Design and Cloning
- Step 1: Target Selection: Identify all constitutive exons within the first 50-75% of the coding sequence (CDS) of your target gene using reference databases (e.g., Ensembl, UCSC Genome Browser).
- Step 2: Candidate gRNA Identification: Use design tools (e.g., Broad Institute's GPP Portal, ChopChop) to scan the selected exon(s). Input the genomic locus and request all possible sgRNAs with an NGG PAM (for SpCas9).
- Step 3: Prioritization: Filter candidates using Table 1 criteria. Select 3-4 top-ranked sgRNAs per gene to account for variable efficiency.
- Step 4: Oligo Design & Cloning: For lentiviral delivery, design oligonucleotides: Forward: 5'-CACCG[N20]-3', Reverse: 5'-AAAC[N20 reverse complement]C-3'. Clone into a BsmBI-cut lentiviral sgRNA expression backbone (e.g., lentiGuide-puro). Transform, sequence-validate plasmids.
The Fate of the Double-Strand Break: Repair Pathways
The outcome of Cas9 cleavage is not a knockout but a DSB, repaired by competing cellular mechanisms. Understanding these pathways is essential for predicting and validating knockout phenotypes.
CRISPR DSB Repair Pathway Decision
Experimental Protocol: Assessing Knockout Efficiency via T7E1 Assay
- Step 1: Genomic DNA Extraction: 72-96 hours post-transfection/transduction, harvest cells. Extract gDNA using a silica-membrane column kit.
- Step 2: PCR Amplification: Design primers ~300-500 bp flanking the target site. Perform PCR using a high-fidelity polymerase.
- Step 3: Heteroduplex Formation: Purify PCR product. Denature and reanneal: 95°C for 10 min, ramp down to 25°C at -0.1°C/sec.
- Step 4: T7 Endonuclease I Digestion: Digest reannealed DNA with T7E1 enzyme (recognizes and cleaves mismatched DNA). Incubate at 37°C for 1 hour.
- Step 5: Analysis: Run digested products on a 2% agarose gel. Cleaved bands indicate presence of indels. Estimate efficiency by band intensity: % Indel = 100 * (1 - sqrt(1 - (b+c)/(a+b+c))), where a is uncut band intensity, b and c are cut band intensities.
Integrating into a Functional Screen: Workflow
A CRISPR knockout screen requires careful integration of gRNA design, delivery, and phenotypic readout.
CRISPR Knockout Screen Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for CRISPR-Cas9 Knockout Screens
| Item | Function & Critical Notes |
|---|---|
| High-Efficiency Cas9 Nuclease | Stable cell line expressing SpCas9 (or other variant) under a constitutive/inducible promoter. Essential for consistent cleavage. |
| Lentiviral sgRNA Backbone | Plasmid with U6-driven sgRNA scaffold, antibiotic resistance (e.g., puromycin), and viral packaging elements. Enables stable integration. |
| Next-Generation Sequencing (NGS) Kit | For deep sequencing of amplified gRNA regions from genomic DNA to quantify abundance pre- and post-selection. |
| T7 Endonuclease I (T7E1) or Surveyor Nuclease | For rapid, gel-based validation of indel formation at target sites. |
| High-Fidelity DNA Polymerase | For error-free amplification of gRNA sequences from genomic DNA during library preparation and validation. |
| Cell Selection Antibiotic | Matched to resistance marker on Cas9 and sgRNA vectors (e.g., blasticidin for Cas9, puromycin for sgRNA). |
| Genomic DNA Extraction Kit | For high-yield, high-purity gDNA from large cell populations, critical for representative NGS library prep. |
| gRNA Design Software | e.g., CRISPick, CHOPCHOP, or EuPaGDT. Incorporates latest efficiency and specificity rules. |
| NGS Analysis Pipeline | e.g., MAGeCK, BAGEL2. Statistically identifies significantly enriched or depleted gRNAs/genes from screen data. |
Within the framework of CRISPR-Cas9 knockout screen principle research, the transition from single-gene interrogation to genome-wide pooled screening represents a paradigm shift in functional genomics. This leap leverages the scalability and precision of CRISPR-Cas9 to systematically probe gene function across the entire genome in a single, integrated experiment. This whitepaper details the core principles, methodologies, and applications of pooled CRISPR screening, providing an in-depth technical guide for researchers and drug development professionals.
Conceptual and Technical Foundations
Traditional single-gene knockout studies, while informative, are inherently low-throughput and fail to capture the complexity of genetic interactions. Pooled screening overcomes this by combining thousands of individual CRISPR guide RNAs (gRNAs) into a single lentiviral library, enabling the transduction of a complex cell population. The core principle involves tracking gRNA abundance over time, often under a selective pressure (e.g., drug treatment, cell viability), to identify genes whose perturbation confers a phenotype. A drop or enrichment of specific gRNAs points to essential genes or genes involved in the selective pathway.
Quantitative Comparison: Single Gene vs. Pooled Screening
The following table summarizes the key differences in scale, design, and output.
| Parameter | Single-Gene Knockout Study | Genome-Wide Pooled CRISPR Screen |
|---|---|---|
| Genetic Targets | One or a few predefined genes | Entire genome (~18,000-20,000 genes) |
| Experimental Scale | Low-throughput, sequential | High-throughput, parallel |
| Library Complexity | Individual constructs | Pooled library (e.g., 3-10 gRNAs/gene) |
| Typical Delivery | Transfection or low-MOI lentivirus | High-coverage lentiviral transduction (MOI~0.3-0.5) |
| Primary Readout | Phenotypic assay per gene | Deep sequencing of gRNA abundance |
| Key Analysis | Direct statistical comparison (e.g., t-test) | Enrichment/depletion statistics (e.g., MAGeCK, DESeq2) |
| Major Cost Driver | Reagent cost per gene | NGS sequencing depth & library cost |
| Time to Data | Weeks to months for a gene set | ~2-4 weeks for whole genome + analysis |
| Primary Output | Definitive conclusion on specific gene(s) | Ranked list of candidate "hit" genes |
Detailed Experimental Protocol for a Genome-Wide CRISPR Knockout Screen
The following protocol outlines the key steps for a typical negative selection (viability) screen.
1. Library Selection and Preparation:
- Select a validated genome-wide CRISPR knockout library (e.g., Brunello, TorontoKO, GeCKO v2). These typically contain 4-10 gRNAs per gene and ~1000 non-targeting control gRNAs.
- Amplify the plasmid library via ultra-deep transformation in bacteria to maintain complexity. Isophenol-chloroform extract high-quality plasmid DNA.
2. Lentivirus Production:
- Co-transfect HEK293T cells (in a 10-layer cell factory or similar) with:
- Library plasmid DNA
- psPAX2 packaging plasmid
- pMD2.G VSV-G envelope plasmid
- Using a transfection reagent like PEI Max.
- Harvest virus-containing supernatant at 48 and 72 hours post-transfection. Concentrate via ultracentrifugation or tangential flow filtration. Titer the virus on the target cell line.
3. Cell Line Transduction and Selection:
- Day 0: Seed Cas9-expressing target cells. The cell line must stably express Cas9 or be transduced to express it prior to the screen.
- Day 1: Transduce cells with the lentiviral library at a low Multiplicity of Infection (MOI = ~0.3) to ensure most cells receive only one gRNA. Include a spinfection step (e.g., 1000 x g, 30-60 min, 32°C) to enhance efficiency.
- Day 2: Replace medium.
- Day 3: Begin puromycin selection (or other appropriate antibiotic) to eliminate untransduced cells. Maintain selection for 5-7 days. This is the T0 timepoint.
4. Screening and Passaging:
- After selection, passage cells continuously for the duration of the experiment (typically 14-28 days, or ~14 population doublings). Maintain a minimum representation of 500 cells per gRNA at all times to prevent stochastic library dropout. This is critical for statistical power.
- Harvest ~50-100 million cells at T0 (immediately post-selection) and at the final T_end timepoint. Pellet, wash with PBS, and store at -80°C for genomic DNA extraction.
5. Genomic DNA Extraction and gRNA Amplification:
- Extract genomic DNA from cell pellets using a large-scale kit (e.g., Qiagen Blood & Cell Culture DNA Maxi Kit). You will need ~200-400 µg of gDNA per sample for good representation.
- Perform a two-step PCR to amplify the integrated gRNA sequences from the genomic DNA and attach Illumina sequencing adapters and sample barcodes. Use a high-fidelity polymerase.
- Purify PCR products via gel extraction or SPRI beads. Quantify by qPCR or bioanalyzer.
6. Next-Generation Sequencing and Data Analysis:
- Pool amplified libraries and sequence on an Illumina HiSeq or NovaSeq platform to achieve deep coverage (aim for >500 reads per gRNA for T0 samples).
- Bioinformatic Analysis:
- Align sequenced reads to the reference gRNA library.
- Count reads per gRNA for T0 and T_end samples.
- Use specialized algorithms (e.g., MAGeCK, BAGEL, CERES) to normalize counts, compare gRNA abundance between timepoints, and rank genes based on statistical significance of depletion/enrichment.
Visualization of Workflows and Pathways
Pooled CRISPR Screen Workflow
Core CRISPR-Cas9 Knockout Mechanism
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Pooled Screening |
|---|---|
| Validated Genome-Wide gRNA Library (e.g., Brunello) | Pre-designed, cloned plasmid pool targeting all human genes with high-efficiency gRNAs and non-targeting controls. Essential for screen integrity. |
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Second/third-generation systems for producing replication-incompetent, high-titer lentivirus to deliver the gRNA library. |
| Cas9-Expressing Cell Line | Target cell line with stable, constitutive Cas9 expression. Critical for efficient and uniform genome editing. |
| Polybrene / Hexadimethrine Bromide | A cationic polymer that enhances viral transduction efficiency by neutralizing charge repulsion between virus and cell membrane. |
| Puromycin (or Blasticidin, etc.) | Selection antibiotic to kill untransduced cells after library delivery, ensuring the population only contains gRNA-bearing cells. |
| High-Fidelity PCR Kit (e.g., KAPA HiFi) | For accurate amplification of gRNA sequences from genomic DNA without introducing bias or errors during library prep for sequencing. |
| NGS Sequencing Platform (Illumina) | Provides the deep, quantitative sequencing required to measure gRNA abundance changes with high accuracy across the complex library. |
| Bioinformatics Pipeline (MAGeCK, BAGEL) | Specialized software to statistically analyze NGS count data, identify significantly enriched/depleted genes, and control for false positives. |
The systematic interrogation of gene function on a genome-wide scale has been a cornerstone of modern biology and drug discovery. The evolution from RNA interference (RNAi) and arrayed screening methods to CRISPR-Cas9-based screening represents a fundamental technological leap, driven by the need for higher specificity, reduced off-target effects, and the ability to model diverse genomic alterations. This transition is central to advancing the thesis that CRISPR-Cas9 knockout screens provide a more precise and comprehensive platform for mapping genotype-to-phenotype relationships, identifying therapeutic targets, and understanding mechanisms of drug action and resistance.
The Pre-CRISPR Era: RNAi and Arrayed Screens
RNA Interference (RNAi) Screening
RNAi utilizes small interfering RNAs (siRNAs) or short hairpin RNAs (shRNAs) delivered via vectors to degrade target mRNA, achieving gene knockdown. Genome-wide libraries target tens of thousands of genes.
Limitations:
- Incomplete Knockdown: Transient reduction, not complete elimination, of gene function.
- High Off-Target Effects: Seed-sequence homology leads to unintended mRNA targeting.
- Cellular Compensation: Phenotypes can be masked by adaptive responses.
Arrayed vs. Pooled Screening Formats
Early functional genomics relied on distinct logistical formats.
Arrayed Screening: Each genetic perturbation (e.g., a single siRNA or cDNA) is delivered into individual wells of a multi-well plate. Phenotypes are measured per well (e.g., high-content imaging, luminescence). Pooled Screening: A heterogeneous library of perturbations (e.g., shRNA or sgRNA vectors) is delivered en masse to a population of cells. Cells are selected based on a phenotype (e.g., drug resistance), and the perturbations conferring the phenotype are identified via next-generation sequencing (NGS) of integrated barcodes.
Table 1: Comparison of Key Pre-CRISPR Screening Modalities
| Feature | Arrayed RNAi | Pooled shRNA | Arrayed cDNA |
|---|---|---|---|
| Perturbation | Knockdown (siRNA) | Knockdown (shRNA) | Overexpression |
| Format | Well-by-well | Pooled | Well-by-well |
| Throughput | High | Very High | Moderate |
| Phenotype Readout | Rich, multivariate | Selective (e.g., survival) | Rich, multivariate |
| Major Limitation | Off-target effects, incomplete knockdown | Off-target effects, false positives | Non-physiological expression |
Protocol: Typical Pooled shRNA Screen
- Library Transduction: A lentiviral shRNA library is transduced into cells at a low MOI to ensure single integration.
- Selection: Cells are selected with puromycin to generate a stable population.
- Phenotype Application: The pool is split, and a selection pressure (e.g., drug treatment) is applied to one arm.
- Harvest & Barcode Amplification: Genomic DNA is harvested from pre-selection and post-selection pools. Integrated shRNA barcodes are PCR-amplified.
- NGS & Analysis: Barcodes are sequenced and counted. Depleted or enriched shRNAs are identified by comparing counts between conditions.
The CRISPR-Cas9 Revolution
The adaptation of the prokaryotic CRISPR-Cas9 immune system for genome engineering enabled permanent, targeted gene knockout via DNA double-strand breaks (DSBs) and error-prone non-homologous end joining (NHEJ). For screening, a single guide RNA (sgRNA) library directs the Cas9 nuclease.
Key Advantages Over RNAi:
- Direct DNA Targeting: Eliminates gene function at the genomic level.
- Higher Specificity: Reduced off-target effects with optimized sgRNA design.
- Multiplexability: Enables combinatorial screening.
- Versatility: Beyond knockout (CRISPRi, CRISPRa, base editing, etc.).
Quantitative Comparison of Screening Technologies
Table 2: Performance Metrics: RNAi vs. CRISPR-KO Screening
| Metric | Pooled shRNA Screening | Pooled CRISPR-KO Screening | Source / Note |
|---|---|---|---|
| Typical Knockdown Efficiency | 70-90% (protein dependent) | ~100% (frameshift mutations) | (Recent reviews, 2023-24) |
| False Positive Rate (Phenotype) | High (Often >10%) | Low (Typically <5%) | (Benchmarking studies) |
| False Negative Rate | High (Due to incomplete knockdown) | Lower (Due to complete knockout) | (Benchmarking studies) |
| Library Size (Human Genome) | ~50,000 shRNAs | ~100,000 sgRNAs | (Brunello, Calabrese libraries) |
| Optimal Screen Duration | 1-2 weeks | 2-4 weeks | (Allows for protein turnover) |
| Typical Pearson Correlation (Replicates) | 0.6-0.8 | 0.85-0.95 | (Experimental data) |
Table 3: Evolution of Screening Capabilities
| Era | Primary Technology | Key Innovation | Major Limitation Addressed |
|---|---|---|---|
| Early 2000s | Arrayed siRNA | High-throughput, single-well readouts | Scalability for complex phenotypes |
| Mid 2000s | Pooled shRNA | Scalability, barcoded NGS readout | Throughput for survival-based screens |
| Early 2010s | Arrayed CRISPR | Precise knockout with HCI compatibility | Throughput and cost |
| Post-2013 | Pooled CRISPR-KO | High-specificity, complete knockout | Specificity and phenotypic penetrance |
| Current (2020s) | CRISPR Perturb-seq (CROP-seq) | Single-cell transcriptomic readout | Molecular phenotype resolution |
Core Protocol: Genome-Wide Pooled CRISPR-Cas9 Knockout Screen
This protocol is fundamental to the thesis on CRISPR-Cas9 knockout screen principle research.
Part 1: Library Design & Preparation
- sgRNA Library Selection: Choose a genome-wide library (e.g., Brunello, with 4 sgRNAs/gene and ~1000 non-targeting controls).
- Library Amplification: Transform the plasmid library into E. coli and culture on large agar plates to maintain representation. Harvest plasmid DNA via maxiprep.
Part 2: Lentiviral Production
- Transfection: Co-transfect HEK293T cells with the sgRNA library plasmid, a psPAX2 packaging plasmid, and a pMD2.G envelope plasmid using PEI transfection reagent.
- Virus Harvest: Collect lentivirus-containing supernatant at 48 and 72 hours post-transfection. Concentrate via ultracentrifugation.
- Titration: Transduce target cells with serial dilutions of virus, then select with puromycin. Calculate titer (TU/mL) based on survival.
Part 3: Screen Execution
- Cell Line Engineering: Generate a Cas9-expressing cell line via lentiviral transduction and blasticidin selection, or use a stable line.
- Library Transduction: Transduce cells at an MOI of ~0.3 to ensure most cells receive one sgRNA. Use a library coverage of >500 cells/sgRNA.
- Selection: Treat with puromycin (for sgRNA vector selection) for 5-7 days.
- Phenotypic Selection: Split cells into experimental (e.g., + drug) and control (e.g., DMSO) arms. Passage cells for 14-21 days, maintaining sufficient coverage.
- Genomic DNA (gDNA) Harvest: Harvest ~1e7 cells per arm. Extract gDNA (e.g., Qiagen Maxi Prep).
Part 4: Sequencing & Analysis
- sgRNA Amplification: Perform two-step PCR on gDNA. PCR1 amplifies the sgRNA region with indexed primers. PCR2 adds Illumina sequencing adapters.
- Next-Generation Sequencing: Pool purified PCR products and sequence on an Illumina platform (MiSeq/HiSeq) to get >500 reads/sgRNA.
- Bioinformatic Analysis:
- Read Alignment: Map reads to the reference sgRNA library.
- Count Normalization: Normalize counts per sample (e.g., counts per million).
- Hit Identification: Use statistical algorithms (MAGeCK, BAGEL) to compare sgRNA abundances between conditions. Genes with significantly depleted or enriched sgRNAs are identified as essential or resistance-conferring, respectively.
Visualizing the Experimental and Conceptual Workflow
Pooled CRISPR-KO Screening Core Workflow
Evolution of Functional Genomics Screening Platforms
The Scientist's Toolkit: Essential Reagents for CRISPR Screening
Table 4: Key Research Reagent Solutions
| Reagent / Material | Function & Description | Example Vendor/Product |
|---|---|---|
| Genome-wide sgRNA Library | Pre-designed, cloned plasmid pool targeting all human/mouse genes with multiple sgRNAs and controls. | Addgene (Brunello, Brie, Mouse Yolk); Dharmacon (Edit-R) |
| Lentiviral Packaging Plasmids | Second-generation system for producing safe, high-titer lentivirus (psPAX2, pMD2.G). | Addgene |
| Cas9-Expressing Cell Line | Stable cell line constitutively expressing SpCas9, eliminating need for co-delivery. | ATCC (e.g., HEK293-Cas9); generated in-house |
| Polybrene / Hexadimethrine Bromide | A cationic polymer that enhances viral transduction efficiency by neutralizing charge repulsion. | Sigma-Aldrich |
| Puromycin Dihydrochloride | Selection antibiotic for cells transduced with puromycin-resistance (PuroR)-bearing sgRNA vectors. | Thermo Fisher Scientific |
| Next-Generation Sequencing Kit | For preparing and sequencing the amplified sgRNA pool from genomic DNA. | Illumina (NovaSeq), Twist Bioscience (NGS reagents) |
| Genomic DNA Extraction Kit | For high-yield, high-quality gDNA extraction from millions of cultured cells. | Qiagen (Blood & Cell Culture DNA Maxi Kit) |
| sgRNA Amplification Primers | Indexed PCR primers designed to specifically amplify the sgRNA cassette from genomic DNA for NGS. | Integrated DNA Technologies (IDT) |
| Bioinformatics Software | Statistical package for analyzing NGS count data to identify significantly enriched/depleted genes. | MAGeCK, BAGEL, CRISPRcleanR |
Within the framework of CRISPR-Cas9 knockout screen principle research, three core concepts are paramount: the design of the gRNA library, the application of selective pressures, and the measurement of phenotypic outcomes. This guide provides a technical dissection of these elements, forming the operational foundation for functional genomics screens aimed at identifying genes essential for specific biological processes or drug responses.
gRNA Library: The Interrogation Toolkit
A gRNA (guide RNA) library is a pooled collection of DNA vectors, each encoding a unique gRNA sequence designed to direct the Cas9 nuclease to a specific genomic target for knockout. The library's composition determines the screen's scope and resolution.
- Genome-Wide vs. Focused Libraries: Genome-wide libraries (e.g., Brunello, Brie) target ~20,000 human genes with 4-10 gRNAs per gene to ensure statistical robustness. Focused libraries target a subset of genes (e.g., kinase family, cancer-associated genes) with higher gRNA density (e.g., 10-20 per gene) for deeper interrogation.
- gRNA Design Principles: Modern libraries are optimized using algorithms that predict on-target efficacy and minimize off-target effects. Key parameters include specific nucleotide compositions (e.g., GC content) and positioning of the seed sequence.
- Library Construction: Libraries are synthesized as oligonucleotide pools, cloned into lentiviral backbone vectors, packaged into virus, and titrated to ensure low Multiplicity of Infection (MOI ~0.3-0.5) to guarantee most cells receive a single gRNA.
Table 1: Common CRISPR Knockout Library Examples
| Library Name | Target Scope | gRNAs per Gene | Approx. Total Size | Primary Use Case |
|---|---|---|---|---|
| Brunello | Human genome-wide | 4 | ~77,000 | High-confidence loss-of-function screens |
| Brie | Human genome-wide | 3 | ~70,000 | Reduced size for higher coverage |
| Mouse Brie | Mouse genome-wide | 3 | ~63,000 | Murine genetic screens |
| Kinase/Phosphatase | Focused (~1,000 genes) | 10-20 | ~10,000 - 20,000 | Signaling pathway dissection |
| Custom Library | User-defined | Variable | Variable | Hypothesis-driven research |
Positive and Negative Selection: Applying Evolutionary Pressure
Selection screens apply environmental pressure to enrich or deplete cells harboring specific gRNAs, revealing gene functions essential for survival under defined conditions.
Positive Selection
Identifies genes whose knockout confers a survival or growth advantage.
- Principle: Under a lethal condition (e.g., toxin, drug, nutrient deprivation), cells with gRNAs targeting essential for condition genes survive and proliferate. These gRNAs are enriched in the final population.
- Common Applications: Identifying drug resistance mechanisms, synthetic lethal interactions, or genes required for pathogen entry.
Negative Selection (Drop-out Screens)
Identifies genes essential for fundamental survival (fitness genes) or for growth under a specific baseline condition.
- Principle: Under normal growth conditions, cells with gRNAs targeting fitness genes are outcompeted and lost. These gRNAs are depleted over time.
- Common Applications: Discovering essential genes for cell proliferation, viability, or housekeeping functions.
Experimental Protocol: Core Screening Workflow
- Cell Line Preparation: Use a Cas9-expressing cell line or co-transduce with Cas9 and the gRNA library.
- Library Transduction: Transduce cells at low MOI (0.3-0.5) to ensure single gRNA integration. Maintain a minimum of 500-1000 cells per gRNA for representation.
- Selection & Passaging: Apply puromycin (for vector selection) for 3-7 days. Split cells into control and experimental arms.
- Pressure Application (T₀): For positive selection, apply the selective agent to the experimental arm. For a negative selection fitness screen, passage both arms under normal conditions for ~14-21 population doublings.
- Harvest Genomic DNA: Collect cells at the initial timepoint (T₀) after selection and at the experimental endpoint (T₁).
- gRNA Amplification & Sequencing: PCR-amplify the gRNA cassette from genomic DNA and perform next-generation sequencing (NGS).
- Data Analysis: Quantify gRNA read counts. Compute log₂ fold-changes (T₁ vs. T₀) and perform statistical analysis (e.g., MAGeCK, CERES) to identify significantly enriched or depleted gRNAs/genes.
Title: CRISPR Knockout Screening Experimental Workflow
Phenotypic Readouts: Measuring the Outcome
The phenotypic readout is the measurable cellular consequence used to score the effect of each knockout.
Table 2: Common Phenotypic Readouts in CRISPR Screens
| Readout Type | Measurement | Screening Format | Key Advantage | Key Limitation |
|---|---|---|---|---|
| Viability/Proliferation | gRNA abundance over time (NGS) | Pooled, Negative Selection | Unbiased, genome-wide, simple | Only measures fitness |
| Drug Resistance | gRNA enrichment post-treatment (NGS) | Pooled, Positive Selection | Directly IDs resistance mechanisms | Requires lethal dose |
| Fluorescence (FACS) | Reporter signal intensity (GFP/RFP) | Pooled or Arrayed | Quantitative, multi-parameter | Throughput limited by sorting |
| Cell Morphology | High-content imaging features | Primarily Arrayed | Rich, multi-feature data | Low throughput, costly |
| Protein Expression | Surface marker (FACS) or barcodes | Pooled (e.g., CITE-seq) | Direct protein-level data | Complex assay setup |
Detailed Protocol: A Positive Selection Drug Resistance Screen
Objective: Identify genes whose knockout confers resistance to Chemotherapy Agent X.
- Day -3: Seed Cas9-expressing cells.
- Day 0: Transduce with genome-wide gRNA library at MOI=0.4. Include a non-targeting control gRNA pool.
- Day 1: Replace virus-containing media.
- Day 3: Begin puromycin selection (2 μg/mL). Maintain for 5 days.
- Day 8 (T₀): Harvest 5e6 cells as the reference timepoint. Extract gDNA (Qiagen Blood & Cell Culture DNA Kit). Freeze pellets for remaining cells.
- Day 8: Split remaining cells into two flasks: Control (DMSO) and Treated (Agent X at IC90). Culture, passaging every 3-4 days, ensuring >500x coverage per gRNA.
- Day 22 (T₁): Harvest all remaining cells (~14 days post-treatment). Extract gDNA.
- NGS Sample Prep: Perform two-step PCR. PCR1: Amplify gRNA region from 10 μg gDNA per sample with indexing primers. PCR2: Add Illumina adapters and sample barcodes. Pool and purify PCR products.
- Sequencing: Run on Illumina NextSeq (75bp single-end). Aim for >300 reads per gRNA.
- Analysis: Align reads to library manifest. Count reads per gRNA in T₀ and T₁ samples. Use MAGeCK algorithm to test for significant enrichment in T₁-treated vs. T₀ or vs. T₁-control. Top hits are candidate resistance genes.
Title: Genetic Mechanism of Drug Resistance in a Positive Selection Screen
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Screen | Critical Considerations |
|---|---|---|
| Cas9-Expressing Cell Line | Provides constant nuclease activity. | Stable, uniform expression is critical; verify editing efficiency before screening. |
| Validated gRNA Library | Contains the pooled genetic perturbations. | Use a recently optimized, published library (e.g., Brunello). Aliquot and store at -80°C. |
| Lentiviral Packaging Plasmids | (psPAX2, pMD2.G) to produce library virus. | Use high-purity endotoxin-free preparations for efficient packaging. |
| Polybrene (Hexadimethrine bromide) | Enhances viral transduction efficiency. | Titrate for each cell line; typical range 4-8 μg/mL. |
| Puromycin (or other antibiotic) | Selects for cells successfully transduced with the library vector. | Determine kill curve for cell line prior to screen; typical range 1-5 μg/mL. |
| Next-Generation Sequencing Kit | (Illumina) to quantify gRNA abundance. | Must be compatible with high-throughput amplicon sequencing. |
| gDNA Extraction Kit | Isolate high-quality, high-molecular-weight gDNA from millions of cells. | Scalability and yield are paramount (e.g., Qiagen Maxi Prep kits). |
| PCR Purification Kit | Clean up amplified gRNA fragments for sequencing. | Minimize bias; use bead-based cleanup for consistency. |
| Bioinformatics Software | (MAGeCK, CRISPRcleanR) to analyze gRNA read counts. | Essential for robust hit calling and correcting for screen-specific biases. |
The integration of a comprehensively designed gRNA library, the strategic application of positive or negative selection, and the precise measurement of a relevant phenotypic readout constitute the methodological triad of a successful CRISPR-Cas9 knockout screen. Mastery of these key definitions and their technical execution enables researchers to systematically decode gene function and identify novel therapeutic targets within complex biological systems.
A Step-by-Step Guide: Designing and Executing a CRISPR-Cas9 Knockout Screen
Within CRISPR-Cas9 knockout (KO) screening research, the foundational step is the precise articulation of the biological question. This determines whether a positive or negative selection screening strategy is appropriate. The choice dictates library design, experimental timeline, and data analysis. Positive selection identifies genes whose loss confers a survival or proliferation advantage (e.g., drug resistance). Negative selection identifies genes essential for survival or proliferation under a given condition, where their loss leads to depletion from the population.
Screening Strategy: A Comparative Framework
The core distinction between positive and negative selection strategies is summarized in the table below.
Table 1: Core Characteristics of Positive vs. Negative Selection CRISPR Screens
| Feature | Positive Selection Screen | Negative Selection Screen |
|---|---|---|
| Biological Question | What gene loss confers a selective advantage? (e.g., resistance to a toxin, growth in low nutrients) | What gene loss causes a fitness defect or lethality? (e.g., essential genes, genes required for pathway activity) |
| Phenotype Measured | Enrichment of sgRNAs/ cells in the treated/selected population vs. control. | Depletion of sgRNAs/ cells in the treated population vs. control. |
| Typical Assay Endpoint | Survival or proliferation under selective pressure. | Relative depletion after a fixed number of cell divisions. |
| Key Analytical Metric | Fold-change enrichment; ranked gene list. | Depletion log2 fold-change; significance (p-value, false discovery rate). |
| Common Applications | Identifying drug resistance mechanisms, synthetic lethal partners, genes allowing survival in stress. | Identifying essential genes, genes required for specific signaling pathways, toxic drug targets. |
| Statistical Power | Higher; focused on strong "hits" that rise above background. | Lower; must distinguish subtle depletion signals from noise; requires greater depth. |
| Library Size & Complexity | Can use genome-wide or focused libraries. | Often uses sub-libraries (e.g., kinase, druggable genome) to maintain high coverage. |
| Timeline | Shorter; selection applied until resistant pools emerge. | Longer; requires multiple population doublings to observe depletion. |
Detailed Experimental Protocols
Protocol for a Genome-wide Positive Selection Screen (e.g., for Drug Resistance)
Aim: To identify genes whose knockout confers resistance to a targeted therapy.
Materials: See "The Scientist's Toolkit" section.
Procedure:
- Library Transduction: Transduce the target cell population (e.g., A549 cancer cells) with a genome-wide CRISPR KO lentiviral library (e.g., Brunello) at a low MOI (~0.3) to ensure most cells receive a single sgRNA. Include a puromycin selection marker.
- Selection and Expansion: Treat transduced cells with puromycin for 5-7 days to select for successfully transduced cells. Expand the population for 10-14 doublings to establish the "T0" or "Reference" population. Harvest 50-100 million cells as a genomic DNA (gDNA) reference.
- Application of Selective Pressure: Split the remaining library pool into replicate treated and untreated control arms. Treat one arm with the drug of interest at a predetermined IC90-IC99 concentration. Maintain the other arm in standard media.
- Outgrowth: Culture both arms, passaging cells as needed, for 14-21 days or until resistant colonies are visibly apparent in the treated arm.
- Harvesting: Harvest all cell populations (T0 reference, final treated pool, final control pool). Isolate gDNA using a large-scale kit (e.g., Qiagen Maxi Prep).
- sgRNA Amplification & Sequencing: Amplify the integrated sgRNA cassettes from gDNA via a two-step PCR. The first PCR (~25 cycles) amplifies the region from bulk gDNA using specific primers. The second PCR (8-12 cycles) adds Illumina sequencing adapters and sample barcodes. Pool PCR products and sequence on an Illumina NextSeq or HiSeq platform to achieve >500x coverage of the library.
- Data Analysis: Align sequences to the reference sgRNA library. Count sgRNA reads in each sample. Normalize counts across samples. Compare normalized sgRNA abundance in the treated vs. control or T0 samples. Rank genes by the enrichment of their targeting sgRNAs using statistical packages like MAGeCK or BAGEL.
Protocol for a Focused Negative Selection Screen (e.g., for Essential Genes in a Pathway)
Aim: To identify genes essential for cell proliferation under basal conditions.
Procedure:
- Library Transduction & Selection: Transduce cells with a focused library (e.g., a kinase library) as in Step 1 of 3.1. Select with puromycin.
- Establish Baseline (T0): Immediately after puromycin selection, harvest a baseline population (50-100 million cells for gDNA).
- Proliferation Phase: Passage the remaining cell pool, maintaining a minimum representation of 500x library coverage at each passage. Culture cells for 14-21 population doublings.
- Harvest Endpoint (T14/T21): Harvest the final cell population.
- Sequencing & Analysis: Perform gDNA extraction, sgRNA amplification, and sequencing as in 3.1. The key difference is in analysis: essential genes are identified by depletion of their targeting sgRNAs in the endpoint (T14/T21) sample compared to the T0 baseline. Use MAGeCK or BAGEL with a negative selection algorithm to rank genes by essentiality score.
Visualizing Screening Strategies and Workflows
Decision Flow for Screen Type Selection
CRISPR Screen End-to-End Experimental Workflow
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for CRISPR-Cas9 Knockout Screens
| Item | Function & Rationale |
|---|---|
| Validated Genome-wide sgRNA Library (e.g., Brunello, GeCKO v2) | A pooled collection of ~4-6 sgRNAs per gene, designed for high on-target knockout efficiency and minimal off-target effects. Provides coverage of the entire genome. |
| Lentiviral Packaging System (e.g., psPAX2, pMD2.G) | Second/third-generation plasmids for producing safe, replication-incompetent lentiviral particles to deliver the sgRNA and Cas9. |
| Stable Cas9-Expressing Cell Line | A cell line with doxycycline-inducible or constitutive expression of Streptococcus pyogenes Cas9. Essential for efficient cutting upon sgRNA delivery. |
| Puromycin or Blasticidin | Selection antibiotics to eliminate untransduced cells, ensuring the screened population contains the sgRNA library. |
| High-Yield gDNA Extraction Kit (e.g., Qiagen Blood & Cell Culture Maxi Kit) | For reliable isolation of microgram to milligram quantities of high-quality genomic DNA from large cell pellets (>50M cells). |
| Herculase II Fusion DNA Polymerase | High-fidelity, high-processivity polymerase for robust and even amplification of sgRNA sequences from complex gDNA samples during PCR1. |
| Illumina-Compatible Indexed Primers | Custom primer sets for PCR2 that add platform-specific adapters and unique dual indices (UDIs) to allow multiplexed, high-depth sequencing. |
| MAGeCK (Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout) | A robust computational pipeline for analyzing both positive and negative selection screens. Handles count normalization, calculates beta scores (enrichment/depletion), and assigns statistical significance. |
Within the broader thesis on CRISPR-Cas9 knockout screen principle research, the selection and sourcing of the guide RNA (gRNA) library represents a critical foundational step. This decision directly impacts the screen's statistical power, biological relevance, and cost. This guide provides an in-depth technical comparison of genome-wide, focused, and custom library designs, detailing current sourcing options, experimental protocols for library validation, and essential research tools.
Library Design Types: A Comparative Analysis
The choice of library scope is dictated by the research hypothesis, budget, and analytical throughput.
Table 1: Comparative Analysis of gRNA Library Types
| Feature | Genome-Wide Library | Focused/Subset Library | Custom Library |
|---|---|---|---|
| Typical Size | 70,000 - 120,000 gRNAs | 1,000 - 10,000 gRNAs | User-defined, 10 - 50,000 gRNAs |
| Target Scope | All annotated protein-coding genes & non-coding regions | Pre-defined gene sets (e.g., kinases, druggable genome) | Investigator-specified genes/regions |
| gRNAs per Gene | 4-10 (common: 4-6) | 5-10 (higher density common) | User-defined (often 5-10) |
| Primary Use | Unbiased discovery, novel gene identification | Hypothesis-driven, pathway analysis, validation | Specialized targets (e.g., specific isoforms, lncRNAs) |
| Cost | High ($3,000 - $8,000) | Moderate ($1,000 - $3,000) | Variable, can be high for novel design |
| Key Advantage | Comprehensive, no prior bias | Higher screening depth, increased statistical power | Complete flexibility, tailored controls |
| Key Challenge | Multiple-testing correction, lower depth per gene | Requires strong prior hypothesis | Design and validation burden |
| Example Vendors | Addgene (Brunello, Brie), Horizon, Synthego | Addgene (Dolcetto, Calabrese), Custom Arrays | Integrated DNA Tech (IDT), Twist Bioscience |
Sourcing and Design Specifications
Libraries are sourced as pooled oligonucleotide pools, typically cloned into lentiviral backbone vectors (e.g., lentiCRISPRv2, lentiGuide-Puro). Key design parameters include:
- On-Target Efficiency: Modern libraries use algorithms like Doench ‘22-Ruleset 3 or CRISPResso2 for prediction. Average predicted efficiency for top libraries exceeds 90%.
- Off-Target Minimization: Designs minimize off-targets with ≤3 mismatches. Specificity scores (e.g., CFD score) are used for filtering.
- Control gRNAs: Essential components include:
- Non-targeting controls (NTCs): 100-1000 gRNAs with no homology to the genome.
- Positive essential gene controls: gRNAs targeting core essential genes (e.g., RPA3, PSMC2) to monitor screen performance.
- Negative safe-harbor controls: gRNAs targeting genomic "safe harbors" (e.g., AAVS1).
Experimental Protocol: Library Cloning and Lentiviral Production
Protocol 1: Cloning of Oligo Pools into Lentiviral Vectors
- Materials: Received oligo pool (desalted, 10-100 ng), BsmBI-v2 digested backbone plasmid (e.g., lentiGuide-Puro, 50 ng/µL), T4 DNA Ligase, Electrocompetent E. coli (e.g., Endura, Stbl4).
- Method:
- Annealing & Phosphorylation: Resuspend oligo pool. Set up annealing reaction: 1 µL oligo pool, 1 µL T4 Ligation Buffer, 7.5 µL nuclease-free water, 0.5 µL T4 PNK. Thermocycler: 37°C 30 min; 95°C 5 min; ramp to 25°C at 5°C/min.
- Golden Gate Cloning: Assemble reaction: 25 ng digested backbone, 0.5 µL annealed oligo (1:200 dilution), 1 µL T4 Ligase, 1 µL BsmBI-v2, 2 µL 10x T4 Buffer, water to 20 µL. Cycle: (37°C, 5 min; 20°C, 5 min) x 30 cycles; then 55°C 5 min, 80°C 5 min.
- Transformation: Desalt ligation with spin column. Electroporate into 25 µL Endura cells (2.5 kV, 1 mm cuvette). Recover in 1 mL SOC for 1 hour at 37°C.
- Plasmid Library Amplification: Plate entire recovery on 5 x 245 mm LB+Amp plates. Incubate 16 hours at 32°C (to prevent recombination). Scrape and maxiprep plasmid DNA. Critical: Ensure library representation >200x colony count per unique gRNA.
Protocol 2: High-Titer Lentivirus Production for Screening
- Materials: Library plasmid DNA, psPAX2 packaging plasmid, pMD2.G envelope plasmid, HEK293T cells, PEI-Max transfection reagent, Lenti-X concentrator.
- Method:
- Seed 15 million HEK293T cells in 15 cm dish 24h pre-transfection (80% confluency).
- For 1 dish: Mix 22.5 µg library plasmid, 16.5 µg psPAX2, 6 µg pMD2.G in 1.5 mL Opti-MEM. In separate tube, mix 112.5 µL PEI-Max in 1.5 mL Opti-MEM. Incubate 5 min.
- Combine DNA and PEI mixes, incubate 20 min at RT. Add dropwise to cells.
- Replace media with 20 mL fresh media 6-8h post-transfection.
- Harvest supernatant at 48h and 72h post-transfection. Pool, filter through 0.45 µm PES filter.
- Concentrate using Lenti-X concentrator (1:3 ratio). Aliquot and titer on target cells (e.g., via puromycin resistance colony formation or qPCR). Aim for titer > 1 x 10^8 TU/mL. Store at -80°C.
Visualization of Key Concepts
gRNA Library Selection and Screening Workflow
gRNA Design and Quality Control Parameters
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for gRNA Library Screening
| Item | Vendor Examples | Function in Experiment |
|---|---|---|
| Validated Genome-Wide Library Plasmid | Addgene (Brunello #73179), Horizon (Dolcetto) | Pre-designed, cloned, and sequence-verified library for immediate virus production. |
| Oligo Pool Synthesis | Twist Bioscience, IDT, Agilent | High-fidelity synthesis of custom gRNA sequence libraries as a single DNA pool. |
| Lentiviral Backbone Vector | Addgene (lentiGuide-Puro #52963, lentiCRISPRv2 #52961) | Receives cloned gRNA pool; contains puromycin resistance for selection. |
| Packaging Plasmids (2nd Gen) | Addgene (psPAX2 #12260, pMD2.G #12259) | Required for production of VSV-G pseudotyped lentiviral particles. |
| High-Efficiency Competent Cells | Lucigen (Endura ElectroCompetent), Thermo Fisher (Stbl4) | Essential for high-complexity library transformation without recombination. |
| Lentiviral Concentration Reagent | Takara Bio (Lenti-X), System Biosciences (PEG-it) | Concentrates low-titer viral supernatant to achieve high MOI stocks. |
| Titer Assay Kit | Takara Bio (Lenti-X qRT-PCR), Abcam (p24 ELISA) | Quantifies functional viral titer before screening to calculate MOI accurately. |
| Next-Gen Sequencing Kit | Illumina (MiSeq Nano, 300-cycle), Custom primers for gRNA amplification | For assessing pre- and post-screen library representation and complexity. |
Within the framework of CRISPR-Cas9 knockout screening for functional genomics and drug target discovery, the delivery of the guide RNA (gRNA) library into the target cell population is a critical determinant of success. Lentiviral transduction remains the gold standard for this step due to its ability to stably integrate into both dividing and non-dividing cells, ensuring permanent gRNA expression. This section details the technical considerations and protocols for executing this phase, with a paramount focus on achieving optimal library coverage to prevent bottlenecking and ensure statistical robustness in screening outcomes.
Core Principle: The Multiplicity of Infection (MOI) and Coverage
The goal is to transduce the cell population such that each cell receives, on average, a single viral integration event. This minimizes the probability of a cell receiving multiple gRNAs, which confounds phenotypic analysis. The key metric is the Multiplicity of Infection (MOI), defined as the ratio of transducing viral particles to target cells. An MOI of ~0.3-0.4 is typically targeted to ensure that most transduced cells receive a single gRNA, following a Poisson distribution.
Library Coverage (C) refers to the number of cells transduced per unique gRNA in the library. To ensure every gRNA is represented adequately in the screened population, a minimum coverage of 200-1000x is recommended. This buffers against stochastic loss and allows for robust statistical power in hit identification.
Quantitative Relationship:
Where the Fraction of transduced cells is determined by the MOI.
Table 1: Key Parameters for Lentiviral Transduction in CRISPR Screens
| Parameter | Recommended Value | Rationale & Calculation |
|---|---|---|
| Target MOI | 0.3 - 0.4 | Ensures >90% of transduced cells receive a single viral integration (Poisson distribution: P(0)=~0.74, P(1)=~0.22, P(>1)=~0.04 at MOI=0.3). |
| Minimum Library Coverage | 200 - 1000x | Provides statistical confidence that each gRNA is represented sufficiently to measure its phenotypic effect. |
| Cell Number for Transduction | (Library Size × Coverage) / Transduction Efficiency | For a 100,000 gRNA library at 500x coverage and 30% transduction efficiency: (100,000 × 500) / 0.3 = ~167 million cells. |
| Viral Titer Requirement | (MOI × Number of Cells) / Viral Volume | To transduce 50M cells at MOI=0.3 with 1 mL of virus: required titer = (0.3 × 50e6) / 1e-3 = 1.5e7 TU/mL. |
| Post-Transduction Selection | Puromycin (1-5 µg/mL) for 3-7 days | Ensures analysis is restricted to successfully transduced, gRNA-expressing cells. |
Table 2: Comparison of Transduction Enhancement Reagents
| Reagent | Mechanism of Action | Typical Use Concentration | Advantages | Considerations |
|---|---|---|---|---|
| Polybrene | Cationic polymer, neutralizes charge repulsion | 4-8 µg/mL | Inexpensive, widely used. | Can be cytotoxic for sensitive cell lines. |
| Hexadimethrine Bromide | Similar to Polybrene | 4-8 µg/mL | Common alternative to Polybrene. | Similar cytotoxicity concerns. |
| Protamine Sulfate | Cationic agent | 4-8 µg/mL | May be less toxic than Polybrene for some cells. | Efficiency varies by cell type. |
| Lentiboost / ViroBoost | Proprietary polymers | As per manufacturer | Often reports higher efficiency & lower toxicity. | Significantly more expensive. |
| Spinoculation | Centrifugation (e.g., 2000 × g, 90 min, 32°C) | N/A | Forces virus-cell contact; can greatly enhance efficiency. | Requires specialized centrifuge with temperature control. |
Detailed Experimental Protocol
Pre-Transduction: Viral Titer Determination (Functional Titering)
Aim: To determine the functional titer (Transducing Units per mL, TU/mL) of your lentiviral gRNA library stock.
Materials: HEK293T or other permissive cells, polybrene, puromycin, growth medium.
Procedure:
- Seed HEK293T cells in a 24-well plate at 50,000 cells/well in 0.5 mL complete medium. Incubate overnight.
- Serially dilute the lentiviral stock (e.g., 10⁻² to 10⁻⁶) in medium containing 8 µg/mL polybrene.
- Remove medium from cells and add 0.5 mL of each virus dilution to duplicate wells. Include a no-virus control with polybrene.
- Incubate for 24 hours, then replace with fresh medium.
- 48 hours post-transduction, split cells and begin selection with puromycin (concentration determined by kill curve).
- After 5-7 days of selection, stain viable colonies with crystal violet or count cells.
- Calculate titer:
TU/mL = (Number of colonies or surviving cells × Dilution Factor) / Volume of virus (mL). Use wells with 20-200 colonies for accuracy.
Main Transduction for Genome-Wide Screen
Aim: To transduce the target cell population at low MOI with high coverage.
Day -1: Cell Preparation
- Harvest exponentially growing target cells.
- Seed the required number of cells (calculated from Table 1) in an appropriate vessel (e.g., 15-cm plates) to reach ~20-30% confluence on the day of transduction. This ensures cells are in log phase and healthy.
Day 0: Viral Transduction
- Prepare Virus-Cell Mix: Thaw viral library aliquot on ice. Pre-warm medium and transduction enhancer (e.g., polybrene at final 8 µg/mL or alternative).
- Mix Calculation: For each replicate, prepare enough virus-cell mix for all plates. Example for one 15-cm plate with 2.5M cells, targeting MOI=0.3 with a viral titer of 1e7 TU/mL:
- Virus Volume (mL) = (MOI × Number of Cells) / Titer = (0.3 × 2.5e6) / 1e7 = 0.075 mL (75 µL).
- Combine virus, polybrene, and pre-warmed medium to a final volume sufficient to cover the plate (e.g., 10 mL for a 15-cm plate).
- Remove the medium from the pre-seeded cells and gently add the virus-medium mixture.
- (Optional but Recommended) Spinoculation: Place plates in a centrifuge with plate carriers. Spin at 800-2000 × g for 60-90 minutes at 32°C. This significantly enhances transduction efficiency.
- Return plates to the 37°C, 5% CO₂ incubator.
- After 6-24 hours, remove the virus-containing medium and replace with fresh, pre-warmed complete medium.
Day 1-2: Begin Selection
- Approximately 48 hours post-transduction, begin antibiotic selection (e.g., puromycin). The exact timing allows for expression of the resistance gene.
- Critical: Perform a pilot kill curve on non-transduced cells beforehand to determine the minimum puromycin concentration that kills all cells within 3-5 days.
- Maintain selection for 5-7 days, passaging cells as needed while maintaining representation (always keep cell numbers far above
Library Size × Coverage).
Day 7+: Harvest for Screening
- After selection is complete and cells are recovering, harvest a representative sample for genomic DNA extraction (Timepoint T0). This serves as the reference for gRNA representation before the screen's selective pressure.
- Proceed with the main screening experiment (e.g., treating with a drug or infection for positive/negative selection).
Visualizations
Diagram 1 Title: CRISPR Screen Lentiviral Transduction Workflow
Diagram 2 Title: gRNA Integration Distribution at Low MOI
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagent Solutions for Lentiviral CRISPR Screen Transduction
| Item | Function / Purpose | Key Considerations |
|---|---|---|
| Lentiviral gRNA Library | Pre-cloned, high-complexity pool of gRNAs targeting the genome. | Ensure titer, complexity, and representation are validated. Store in small single-use aliquots at -80°C. |
| High-Quality Packaging Plasmids | psPAX2 (gag/pol/rev) and pMD2.G (VSV-G envelope) for virus production. | Use endotoxin-free plasmid preps for higher titer production. |
| Polybrene or Equivalent | Cationic transduction enhancer; increases viral attachment. | Titrate for cytotoxicity. Can use protamine sulfate or commercial boosters as alternatives. |
| Puromycin Dihydrochloride | Selective antibiotic for cells expressing the puromycin resistance gene (PuroR) from the lentiviral vector. | Perform a kill curve on target cells to determine the minimal effective concentration (typically 1-5 µg/mL). |
| Hexadimethrine Bromide | Alternative cationic polymer to Polybrene. | Sometimes reported as less toxic for sensitive cell lines. |
| Lenti-X Concentrator | Chemical concentrator (PEG-it) to increase viral titer if needed. | Useful for low-titer supernatants. Follow protocol to avoid pellet loss. |
| Poly-L-lysine | Coats cultureware to enhance cell adhesion, critical during spinoculation. | Use for poorly adherent cell lines to prevent detachment during centrifugation. |
| Crystal Violet Solution | For staining and quantifying colonies in titering assays. | 0.5-1% in methanol or ethanol. |
| DNase I | Used during viral prep to remove contaminating plasmid DNA, ensuring functional titer reflects true viral particles. | Critical for accurate titer determination. |
Within the broader thesis on CRISPR-Cas9 knockout screen principles, Step 4 represents the critical translational pivot from genetic perturbation to phenotypic discovery. Following library transduction and guide RNA (gRNA) integration, this phase involves subjecting the engineered cell population to a defined environmental challenge—selective pressure—to enrich for cells harboring gRNAs targeting genes essential for survival or proliferation under those conditions. The subsequent harvesting and preparation of samples for sequencing-based deconvolution is a determinant of screen success. This guide details contemporary protocols, data handling, and logistical considerations for executing this pivotal step.
Principles of Selective Pressure Application
The nature of the selective pressure is dictated by the biological question. Common modalities include:
- Viability/Proliferation Screens: Application of cytotoxic compounds (e.g., chemotherapeutics, targeted inhibitors) or culture in nutrient-depleted media to identify genes conferring resistance or sensitivity.
- Fitness Screens: Continuous passaging over multiple cell doublings to identify genes essential for core cellular fitness.
- Signal Transduction Screens: Stimulation with growth factors, cytokines, or other ligands to dissect pathway dependencies.
- Genetic Interaction Screens: Combining CRISPR knockout with a second perturbation (e.g., drug, another genetic alteration) to identify synthetic lethal or rescuing interactions.
The duration of pressure must be optimized to allow sufficient phenotypic divergence between positively and negatively selected gRNA populations, typically spanning 7-21 population doublings.
Quantitative Framework for Pressure Duration & Sampling
Optimal screening parameters are derived from pilot experiments. Key quantitative benchmarks are summarized below.
Table 1: Key Quantitative Benchmarks for Selective Pressure
| Parameter | Typical Range / Target | Measurement Purpose & Rationale |
|---|---|---|
| Cell Coverage (Library Level) | >500x | Ensures each gRNA is represented in sufficient starting copies to mitigate stochastic dropout. |
| MOI (Infection) | 0.3 - 0.4 | Maximizes percentage of cells with a single gRNA integration. |
| Selection Efficiency (Post-Puromycin) | >90% | Validates successful antibiotic selection of transduced cells before applying experimental pressure. |
| Population Doublings under Pressure | 7 - 14 | Balances signal (enrichment/depletion) development with library complexity maintenance. |
| Minimum Fold-Change for Hit Calling | ||
| - Depletion (Essential Gene) | < 0.5 | Commonly used threshold in robust rank aggregation or MAGeCK analyses. |
| - Enrichment (Resistance Gene) | > 2.0 | Identifies gRNAs significantly increased in abundance post-selection. |
| Sequencing Depth per Sample | 50 - 100x read coverage per gRNA | Ensures accurate quantification of gRNA abundance distribution. |
Experimental Protocol: Applying Pressure and Harvesting Genomic DNA
A. Pre-Pressure Preparation
- Cell Expansion: Following puromycin selection, expand cells to the required number for the screen, maintaining a minimum of 500 cells per gRNA in the library.
- Baseline (T0) Harvest: Pellet and freeze a minimum of 20 million cells (or equivalent DNA yield) as the T0 reference time point. Store at -80°C.
- Seeding for Selection: Seed replicate cell populations (technical replicates are critical) at appropriate density into culture vessels for the applied pressure condition(s) and a no-pressure control condition.
B. Applying Selective Pressure
- Initiation: Introduce the selective agent (drug, media change, etc.) to experimental arms. Maintain control populations in standard culture conditions.
- Monitoring: Passage cells as needed, maintaining minimum coverage. Monitor cell count and viability. Document population doublings.
- Duration: Continue pressure for the predetermined number of population doublings (e.g., 10 doublings).
C. Harvesting Samples for gRNA Recovery
- Termination: At endpoint, harvest all cells (control and selected populations) by trypsinization or scraping.
- Cell Counting: Perform accurate cell counts for each sample.
- Cell Pelletting: Pellet 10-20 million cells per sample (or the entirety of smaller populations). Wash once with PBS.
- Genomic DNA (gDNA) Extraction:
- Use a scalable gDNA extraction kit (e.g., Qiagen Blood & Cell Culture DNA Maxi Kit) suitable for high yield and purity.
- Follow manufacturer protocol for cell pellets. Ensure complete cell lysis.
- Elute DNA in a low-EDTA TE buffer or nuclease-free water. Quantify using a fluorometric method (e.g., Qubit).
- Yield Target: Aim for >50 µg of gDNA per 10 million cells as a benchmark.
- Storage: Store gDNA at -20°C or -80°C until PCR amplification.
D. gDNA Amplification & Sequencing Library Prep This protocol is adapted from standard pooled-library amplification methods.
- Primary PCR (Amplify Integrated gRNA Loci):
- Reaction Setup: For each sample, set up multiple 50-100 µL PCR reactions using a high-fidelity polymerase to minimize bias. Use ~5 µg of gDNA total per sample, distributed across reactions.
- Primers: Use forward primers binding the constant region of the lentiviral vector upstream of the gRNA scaffold and reverse primers binding the downstream constant region. Incorporate partial Illumina adapter sequences.
- Cycling Conditions: [98°C 30s] x 1; [98°C 10s, 60°C 15s, 72°C 30s] x 18-22 cycles; [72°C 2 min] x 1. Keep cycles low to limit skew.
- Pool & Purify: Pool all primary PCR reactions for a given sample. Purify using a size-selection magnetic bead clean-up (e.g., SPRIselect beads).
- Secondary PCR (Add Full Sequencing Adapters & Indices):
- Use 5 µL of purified primary PCR product as template.
- Use full-length Illumina indexed primers.
- Run 8-12 cycles.
- Final Purification & Quantification: Purify final libraries, validate size (~250-300 bp) by bioanalyzer, and quantify by qPCR for accurate pooling.
- Sequencing: Pool libraries equimolarly and sequence on an Illumina platform (e.g., NextSeq 500/2000), aiming for 50-100x coverage per gRNA.
Signaling Pathways & Experimental Workflow
Workflow for Selective Pressure & Sample Harvest
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Research Reagent Solutions for Step 4
| Item | Function & Rationale |
|---|---|
| Selective Agent | The chemical, biological, or environmental perturbation (e.g., targeted inhibitor, chemotherapeutic, cytokine) used to challenge the cell population and induce phenotypic selection. |
| Puromycin Dihydrochloride | Selective antibiotic used prior to Step 4 to eliminate non-transduced cells, ensuring a pure population of CRISPR-modified cells for the screen. |
| High-Yield gDNA Extraction Kit (Midi/Maxi Scale) | Scalable kits (e.g., from Qiagen, Thermo Fisher) are essential for obtaining sufficient, high-quality genomic DNA from 10-100 million cells for subsequent PCR. |
| Magnetic Bead-based Purification Kit (e.g., SPRIselect) | For size-selective cleanup and concentration of PCR amplicons, ensuring removal of primers, dimers, and salts before sequencing. |
| High-Fidelity PCR Polymerase (e.g., KAPA HiFi, Q5) | Minimizes amplification bias during gRNA library PCR, crucial for accurate representation of gRNA abundance. |
| Dual-Indexed Illumina PCR Primers | Adds unique sample indices (i7, i5) and full sequencing adapters during secondary PCR, enabling multiplexed sequencing. |
| Fluorometric DNA Quantitation Kit (e.g., Qubit dsDNA HS) | Accurate quantification of low-concentration DNA (gDNA, PCR libraries) without interference from RNA or salts, critical for pooling. |
| Cell Culture Reagents & Vessels | Scalable flasks, plates, and media for maintaining high-coverage cell populations over extended culture periods. |
Within CRISPR-Cas9 pooled knockout screens, quantifying guide RNA (gRNA) abundance before and after a selection pressure is fundamental to identifying genes essential for a given phenotype. Next-Generation Sequencing (NGS) is the enabling technology for this high-throughput quantification. This step involves preparing a sequencing library from the amplified gRNA cassettes extracted from the screen and subsequently using bioinformatic tools to quantify each gRNA's representation. This guide details the current best practices for NGS library preparation and gRNA abundance analysis, critical for the success of the broader screen.
Core Principles of NGS Library Preparation for gRNA Reads
The goal is to convert the PCR-amplified gRNA inserts from the mammalian vector into a format compatible with your NGS platform (e.g., Illumina). This involves adding platform-specific adapter sequences and sample indices (barcodes) to allow multiplexing.
Key Considerations:
- Amplification Bias: Minimizing PCR cycles during library amplification is crucial to prevent skewing gRNA representation.
- Dual Indexing: Using unique dual indices (i-index and p7 index) per sample increases multiplexing capacity and reduces index hopping errors.
- Read Length: A single-end 75-150 bp read is typically sufficient to sequence the constant regions flanking the variable 20bp gRNA sequence.
Detailed Experimental Protocol
Materials and Equipment
| Item | Function/Description |
|---|---|
| PCR-amplified gRNA pool | Input DNA containing the variable gRNA sequences flanked by constant regions. |
| Indexed Illumina P5/P7 Primers | Primer mix containing the universal adapter sequences and unique dual indices for multiplexing. |
| High-Fidelity DNA Polymerase | e.g., KAPA HiFi or Q5. Essential for accurate, low-bias amplification. |
| SPRI Beads | (e.g., AMPure XP) For size selection and cleanup of PCR products, removing primers and primer dimers. |
| Qubit Fluorometer & dsDNA HS Assay Kit | For accurate quantification of library concentration. |
| Bioanalyzer or TapeStation | For assessing library fragment size distribution and quality. |
| Illumina-Compatible Sequencing Kit | e.g., MiSeq Reagent Kit v3 (150-cycle) for quality control sequencing. |
Step-by-Step Workflow
- Dilution & Normalization: Dilute the initial PCR-amplified gRNA pool to a uniform concentration (e.g., 10 ng/µL) across all samples.
- Library PCR (Indexing PCR):
- Set up a 50 µL reaction:
- 25 µL 2X High-Fidelity PCR Master Mix
- 2.5 µL Forward Primer (P5 adapter + i5 index)
- 2.5 µL Reverse Primer (P7 adapter + i7 index)
- 20 µL diluted gDNA/PCR product (≤ 100 ng total)
- Cycling Conditions:
- 98°C for 45 s (initial denaturation)
- 8-12 cycles of: 98°C for 15 s, 60°C for 30 s, 72°C for 30 s
- 72°C for 1 min (final extension)
- Hold at 4°C.
- Critical: Use the minimum cycle number that yields sufficient product (~200 ng total) to minimize bias.
- Set up a 50 µL reaction:
- SPRI Bead Cleanup: Perform a double-sided size selection (e.g., 0.6x ratio to remove large fragments, then 1.2x ratio on the supernatant to recover fragments >150 bp) to purify the final library and remove primer dimers.
- Library Quantification & QC:
- Quantify using Qubit (dsDNA HS assay).
- Analyze size distribution and purity using Bioanalyzer (High Sensitivity DNA chip). Expect a single peak at the expected size (~200-300 bp depending on vector design).
- Pooling & Normalization for Sequencing: Precisely quantify each indexed library by qPCR (e.g., using KAPA Library Quantification Kit) for accurate molarity. Pool libraries at equimolar ratios.
- Sequencing: Sequence on an appropriate Illumina platform. For a typical screen with 1000 gRNAs, a MiSeq run provides sufficient depth for QC. For full-scale screens, a HiSeq or NovaSeq is required. Aim for a minimum of 200-500 reads per gRNA.
gRNA Abundance Quantification & Data Processing
The raw sequencing data (FASTQ files) must be processed to extract gRNA counts.
Title: Bioinformatics Pipeline for gRNA Read Counting
Detailed Protocol for Data Analysis
- Demultiplexing: Use
bcl2fastq(Illumina) to generate per-sample FASTQ files based on the dual indices. Quality Trimming & Adapter Removal: Use
TrimmomaticorCutadapt.- Example Cutadapt command:
- Example Cutadapt command:
Alignment to gRNA Reference Library: Align reads to a FASTA file of all expected gRNA sequences (constant regions + variable 20bp).
- Example Bowtie2 command for an end-to-end alignment:
- Example Bowtie2 command for an end-to-end alignment:
gRNA Read Counting: Count the number of reads aligning uniquely to each gRNA sequence using tools like
featureCounts(from Subread package) or a custom script.- Example featureCounts command:
- Example featureCounts command:
Generation of Count Table: The output is a count matrix with rows as gRNAs and columns as samples (e.g., T0 plasmid, T0 cells, Treated cells).
Key Metrics and Quality Control
Essential QC parameters to assess before proceeding to statistical analysis.
| Metric | Target/Threshold | Purpose/Rationale |
|---|---|---|
| Total Reads per Sample | > 10 million (screen-dependent) | Ensures sufficient sampling depth. |
| Alignment Rate | > 90% | Indicates specificity of library prep and sequencing. |
| Reads Assigned to gRNAs | > 80% of aligned reads | Measures efficiency of gRNA capture. |
| gRNAs Detected | > 95% of library | Assesses library completeness and PCR bias. |
| PCR Bottleneck Coefficient | < 0.5 (calculated pre/post amplification) | Quantifies amplification noise introduced during library prep. |
| Replicate Correlation (R²) | > 0.95 (for technical replicates) | Assesses reproducibility of the NGS process. |
Title: NGS Library Quality Control Decision Tree
The Scientist's Toolkit: Essential Reagents & Materials
| Item | Specific Product Examples (Research-Use Only) | Primary Function |
|---|---|---|
| Library Prep Kit | Illumina DNA Prep Kit | Provides a streamlined, bead-based workflow for adapter ligation and PCR. |
| Indexing Primers | Illumina CD Indexes | Sets of unique dual index primers for multiplexing up to 384 samples. |
| High-Fidelity Polymerase | KAPA HiFi HotStart ReadyMix | Provides high fidelity and yield during the indexing PCR, minimizing bias. |
| Size Selection Beads | SPRIselect / AMPure XP Beads | Magnetic beads for reproducible size selection and cleanup of DNA fragments. |
| Library Quant Kit | KAPA Library Quantification Kit (qPCR) | Enables accurate, molar-based quantification of sequencing libraries. |
| QC Instrument | Agilent 4200 TapeStation | Provides fast, automated analysis of library fragment size and integrity. |
| Alignment Software | Bowtie2 | Fast and memory-efficient aligner for mapping gRNA reads to a reference. |
| Counting Software | MAGeCK | Specifically designed end-to-end tool for CRISPR screen count processing and statistical analysis. |
CRISPR-Cas9 knockout screening has evolved from a foundational genetic tool into a cornerstone of functional genomics. The core thesis of this research domain posits that systematic, genome-wide perturbation enables the quantitative mapping of gene function onto phenotypic outcomes, revealing fundamental biological principles and direct paths to therapeutic intervention. This whitepaper elaborates on two critical validations of this thesis: the definitive identification of context-specific essential genes and the systematic dissection of drug resistance mechanisms.
Essential Gene Identification: Defining Cellular Fitness
The principle that knocking out essential genes leads to loss of cellular fitness is leveraged in negative selection screens. The experimental workflow is designed to identify genes whose loss impairs survival or proliferation.
2.1 Experimental Protocol for a Genome-Wide Negative Selection Screen
- Library Design & Cloning: A genome-wide lentiviral sgRNA library (e.g., Brunello, TKOv3) is used. Each gene is targeted by 4-6 sgRNAs, with ~1000 non-targeting controls.
- Viral Production & Cell Transduction: Produce lentivirus from the library plasmid pool. Transduce target cells at a low Multiplicity of Infection (MOI ~0.3) to ensure most cells receive a single sgRNA. Maintain >500x library representation.
- Selection & Passaging: Apply puromycin (2 µg/mL, 48-72h) to select transduced cells. Harvest an initial reference sample (T0). Passage the remaining population for 14-21 cell doublings, maintaining representation.
- Genomic DNA Extraction & Sequencing: Harvest endpoint samples (Tend). Extract gDNA (Qiagen Blood & Cell Culture DNA Kit). Amplify integrated sgRNA sequences via PCR using indexing primers for NGS.
- Data Analysis: Sequence reads are aligned to the library reference. sgRNA depletion/enrichment is calculated using tools like MAGeCK or CERES, which compare sgRNA abundance at T0 vs. Tend, accounting for copy-number effects and screen quality.
Table 1: Representative Data from a Cancer Cell Line Essential Gene Screen
| Gene | Function | Avg. log2 fold-change (Tend/T0) | FDR-adjusted p-value | Classification |
|---|---|---|---|---|
| PCNA | DNA replication | -4.67 | 2.1E-12 | Core Essential |
| KRAS | Oncogenic driver | -3.21 | 5.8E-09 | Context-Essential |
| CDK4 | Cell cycle kinase | -2.95 | 1.3E-07 | Context-Essential |
| MYH9 | Cytoskeletal motor | -0.12 | 0.84 | Non-essential |
Uncovering Drug Resistance Mechanisms
Positive selection screens identify genes whose knockout confers a survival advantage under selective pressure, such as anti-cancer therapeutics.
3.1 Experimental Protocol for a Drug Resistance Screen
- Library & Cell Line: A targeted library focusing on chromatin modifiers, kinases, or known cancer genes is often used. A drug-sensitive cell line is selected.
- Transduction & Selection: Follow steps 1-3 from Section 2.1. After puromycin selection, split cells into two arms: Drug Treatment and Vehicle Control (DMSO).
- Application of Selective Pressure: Treat cells with the drug at a pre-determined IC70-IC90 concentration. Refresh drug/vehicle media every 3-4 days.
- Harvesting & Sequencing: Harvest treatment and control arms once the control arm has been passaged equivalently to the drug arm (e.g., ~14 doublings) or when resistant clones emerge in the drug arm. Process for NGS as in Step 4, Section 2.1.
- Data Analysis: Use MAGeCK-RRA or similar to identify sgRNAs significantly enriched in the drug-treated arm versus the control arm. Top hits reveal genes whose loss confers resistance.
Table 2: Example Hits from a PARP Inhibitor (Olaparib) Resistance Screen in BRCA1-Mutant Cells
| Gene | Known Function | Avg. Fold-Enrichment (Drug/Control) | FDR p-value | Proposed Resistance Mechanism |
|---|---|---|---|---|
| 53BP1 | DNA repair factor | 45.2 | 4.5E-14 | Loss restores error-prone DSB repair, bypassing HR deficiency. |
| REV7 | Shieldin complex | 38.7 | 9.2E-13 | Loss of shieldin restores end-resection and microhomology-mediated repair. |
| RIF1 | Shieldin complex | 35.1 | 3.1E-12 | Same as REV7. |
| PARP1 | Target of drug | 0.8 | 0.91 | (Negative control, essential for drug efficacy) |
Visualizing Core Concepts and Pathways
Diagram 1: CRISPR Screen Workflow for Fitness & Resistance
Diagram 2: PARPi Resistance via 53BP1/Shieldin Loss
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent/Material | Supplier Examples | Critical Function in Screen |
|---|---|---|
| Genome-wide sgRNA Library (e.g., Brunello, TKOv3) | Addgene, Sigma-Aldrich | Provides comprehensive, validated targeting of all human genes with multiple sgRNAs/gene. |
| Lentiviral Packaging Mix (psPAX2, pMD2.G) | Addgene | Essential for producing high-titer, replication-incompetent lentiviral particles. |
| Polybrene (Hexadimethrine Bromide) | Sigma-Aldrich | Enhances viral transduction efficiency by neutralizing charge repulsion. |
| Puromycin Dihydrochloride | Thermo Fisher, Sigma-Aldrich | Selects for cells successfully transduced with the lentiviral sgRNA construct. |
| Next-Generation Sequencing Kit (Illumina) | Illumina | Enables high-throughput quantification of sgRNA abundance from genomic DNA. |
| MAGeCK Software Suite | Open Source | Standard computational pipeline for robust identification of enriched/depleted sgRNAs from NGS data. |
| Cell Viability Assay Kit (e.g., CellTiter-Glo) | Promega | Used pre-screen to determine optimal drug concentration (ICxx) for positive selection. |
Troubleshooting CRISPR Screens: Addressing Common Pitfalls and Advanced Optimization
Within the broader thesis on CRISPR-Cas9 knockout screen principle research, the reliability and interpretability of screening data are paramount. Three pervasive technical challenges—low infection efficiency, off-target effects, and screen noise—consistently compromise data integrity. This whitepaper provides an in-depth technical guide to understanding, quantifying, and mitigating these issues to ensure robust functional genomics screening.
Low Infection Efficiency
Infection efficiency refers to the percentage of target cells that successfully receive and express the CRISPR-Cas9 components. Low efficiency (<70% for pooled screens) creates a mixed population of edited and unedited cells, diluting phenotypic signals and increasing screen noise.
Table 1: Common Factors Affecting Lentiviral Infection Efficiency
| Factor | Typical Impact Range | Optimal Condition / Mitigation |
|---|---|---|
| Target Cell Type | Primary cells: 10-40%; Immortalized lines: 60-90% | Use early-passage, actively dividing cells. |
| Multiplicity of Infection (MOI) | High MOI (>3) increases risk of multiple integrations. | Aim for MOI of 0.3-0.6 to ensure most cells get a single guide. |
| Polybrene Concentration | 4-8 µg/ml can improve efficiency 1.5-3x for adherent lines. | Titrate for cell type; toxic for some sensitive lines. |
| Spinoculation | Can improve efficiency 2-5x for refractory cells. | 2000 x g, 32°C, 60-120 minutes. |
| Transduction Enhancers (e.g., LentiBoost, Hexadimethrine bromide variants) | Can improve 2-10x for difficult cells (e.g., macrophages, T cells). | Must be titrated to avoid cytotoxicity. |
Detailed Protocol: Determining Functional Titer and MOI
Objective: To establish the viral titer that yields optimal infection with minimal multiple integrations.
- Day 1: Seed 2 x 10⁵ target cells per well in a 12-well plate.
- Day 2: Prepare serial dilutions of the lentiviral guide RNA (gRNA) library stock (e.g., 1:10, 1:100, 1:1000) in complete medium with 8 µg/ml polybrene.
- Replace cell medium with 1 ml of diluted virus. Include a polybrene-only control.
- Spinoculate at 2000 x g, 32°C for 90 minutes. Then, incubate at 37°C.
- Day 3: Replace with fresh complete medium.
- Day 5 (72h post-infection): Harvest cells and analyze by flow cytometry for the expression of a co-delivered marker (e.g., GFP, puromycin resistance via survival assay).
- Calculation: Functional titer (TU/ml) = (Cell number at transduction * % positive cells * dilution factor) / volume of virus (ml). Select the dilution yielding 20-40% positivity for MOI ~0.3-0.6.
Off-Target Effects
Off-target effects occur when Cas9 cleaves genomic sites with sequence homology to the intended gRNA, leading to confounding phenotypes unrelated to the target gene's knockout.
Table 2: Strategies for Off-Target Assessment and Mitigation
| Strategy | Principle & Data Impact | Typical Reduction in Off-Targets |
|---|---|---|
| High-Fidelity Cas9 Variants (e.g., SpCas9-HF1, eSpCas9) | Engineered to reduce non-specific DNA binding. | 2- to 10-fold reduction detectable by GUIDE-seq. |
| Truncated gRNAs (tru-gRNAs) | Using 17-18nt spacers instead of 20nt reduces tolerance to mismatches. | Up to 5,000-fold reduction for some off-target sites. |
| Paired Nickases (Cas9n) | Requires two adjacent off-target sites for a double-strand break. | Can reduce off-target indels to near-background levels. |
| Chemically Modified gRNAs | 2'-O-methyl-3'-phosphonoacetate modifications enhance specificity. | Reported 10- to 100-fold reduction in specific contexts. |
| Bioinformatic gRNA Design | Algorithms (e.g., CHOPCHOP, CRISPOR) score and exclude guides with predicted off-targets. | Minimizes but does not eliminate risk; essential first step. |
Detailed Protocol: Off-Target Validation via GUIDE-seq
Objective: To empirically identify genome-wide off-target sites for a given gRNA.
- Design: Synthesize the GUIDE-seq Oligonucleotide (a 34-bp double-stranded phosphorothioate-modified DNA tag).
- Transfection: Co-transfect 2 x 10⁵ HEK293T cells with 100ng of Cas9 expression plasmid, 50ng of gRNA expression plasmid, and 100pmol of GUIDE-seq oligonucleotide using a high-efficiency transfection reagent.
- Genomic DNA Extraction: Harvest cells 72h post-transfection. Extract gDNA using a silica-column method.
- Library Preparation: Shear gDNA to ~500bp. End-repair, A-tail, and ligate with annealed adaptors containing partial Illumina sequences. Perform a first PCR (15 cycles) with primers specific to the adaptors and the integrated GUIDE-seq tag.
- Target Enrichment & Sequencing: Run a nested, indexed PCR (25 cycles) on the first PCR product. Purify and pool libraries for paired-end sequencing on an Illumina MiSeq or HiSeq.
- Bioinformatic Analysis: Use the GUIDE-seq computational pipeline to align reads, detect tag integrations, and identify off-target sites. Sites with ≥2 unique tag integrations are typically considered valid.
Diagram Title: GUIDE-seq Experimental Workflow for Off-Target Detection
Screen Noise
Screen noise encompasses technical and biological variability that obscures the true phenotype of a gene knockout, leading to false positives and negatives. Key sources include gRNA library design, uneven representation, and batch effects.
Table 3: Sources of Screen Noise and Mitigation Metrics
| Noise Source | Impact Measurement | Recommended Threshold / Mitigation |
|---|---|---|
| Uneven gRNA Representation | Skew in pre-screen read count distribution. | >90% of gRNAs within 10-fold of median read count. |
| PCR Duplication in NGS | Overestimation of gRNA abundance. | Deduplicate based on unique molecular identifiers (UMIs). |
| Batch Effects | Significant difference (p<0.01, Mann-Whitney) in control gRNA distributions between batches. | Normalize using robust z-score or RRA across batches. |
| Copy Number Effects | False positives in essential gene calls in aneuploid regions. | Use CN-correcting algorithms (e.g., CERES, BAGEL2). |
| Variable Knockout Efficacy | In-frame mutation rate leading to escape. | Design 4-6 gRNAs/gene; use algorithms favoring on-target activity. |
Detailed Protocol: Screen De-noising with Control gRNAs
Objective: To normalize screening data and reduce false discoveries using non-targeting and essential gene controls.
- Library Design: Include a minimum of 100 non-targeting control (NTC) gRNAs and 50 gRNAs targeting core essential genes (e.g., from the Hart et al. list) spread across the library.
- Sequencing & Quantification: Sequence the library plasmid pool (pre-screen reference) and genomic DNA from the screen end-point. Align reads, count gRNAs, and calculate read counts per million (RPM).
- Calculate Enrichment Score: For each gRNA i, compute a log2 fold change (LFC): LFCi = log2(RPMpost-screeni / RPMpre-screen_i).
- Normalize Using Controls:
- For Essentiality Screens (Negative Selection): Center the LFC distribution so that the median LFC of NTCs is 0.
- For Enrichment Screens (Positive Selection): Use the median absolute deviation (MAD) of NTCs to compute a robust z-score.
- Gene-Level Scoring: Use the median LFC of all gRNAs targeting a gene, or advanced algorithms like MAGeCK or CRISPRcleanR, which incorporate control gRNAs to model and subtract noise.
- Hit Calling: A gene is a high-confidence hit if it passes a false discovery rate (FDR) threshold (e.g., <5%) and its phenotype is consistent across multiple gRNAs.
Diagram Title: Core Workflow for CRISPR Screen & Noise Reduction
The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions
| Item | Function & Rationale |
|---|---|
| High-Titer Lentiviral Packaging Mix (e.g., psPAX2, pMD2.G) | Produces high-viral-titer supernatants crucial for achieving high infection efficiency in difficult cells. |
| Polybrene or LentiBoost | Cationic polymers that neutralize charge repulsion between virus and cell membrane, enhancing transduction. |
| Puromycin Dihydrochloride | Selection antibiotic for cells transduced with puromycin resistance-containing vectors; critical for eliminating uninfected cells. |
| High-Fidelity Cas9 Plasmid (e.g., pX458-HF1) | Expresses a specificity-enhanced Cas9 variant to mitigate off-target effects in arrayed or low-complexity screens. |
| Validated Control gRNA Plasmids (Non-targeting & Essential) | Essential for normalizing screen data and assessing screen quality. |
| Unique Molecular Identifier (UMI) Adapter Kit (for NGS) | Allows accurate deduplication of PCR amplicons, eliminating noise from PCR amplification bias. |
| Robust Cell Viability Assay (e.g., CellTiter-Glo) | For arrayed screens, provides luminescence-based viability readout with high signal-to-noise. |
| Genomic DNA Cleanup Kit (Silica-column based) | High-yield, pure gDNA is critical for unbiased PCR amplification of gRNA loci during screen deconvolution. |
| Next-Generation Sequencing Kit (Illumina-compatible) | Required for deep sequencing of the gRNA library pre- and post-screen. |
| Bioinformatics Software (MAGeCK, CRISPResso2) | Open-source tools essential for quantifying gRNA abundance, calculating phenotypes, and analyzing editing efficiency. |
CRISPR-Cas9 knockout screens are a cornerstone of functional genomics, enabling genome-wide interrogation of gene function. The core principle involves delivering a library of single guide RNAs (sgRNAs) into cells expressing Cas9 to generate targeted knockouts. The success of these screens is fundamentally dependent on the performance of each individual sgRNA. Therefore, optimizing gRNA design for maximal on-target efficiency and accurate efficacy prediction is critical for achieving high signal-to-noise ratios, reducing false positives/negatives, and ensuring robust biological conclusions.
Core Determinants of gRNA Efficacy
Sequence-Based Features
gRNA efficacy is influenced by specific nucleotide preferences and local sequence context.
Table 1: Key Nucleotide Features Influencing gRNA Cleavage Efficiency
| Feature | Optimal Characteristic | Reported Impact on Efficacy | Biological Rationale |
|---|---|---|---|
| GC Content | 40-60% | High correlation (R≈0.3-0.4) with efficiency | Influences DNA melting and complex stability |
| Positional Nucleotides (PAM Proximal) | 'G' at position 20, 'G' or 'C' at position 19 | Can increase efficiency by up to 2-fold | Affects Cas9 binding and R-loop initiation |
| Thermodynamic Stability (5' end) | Lower stability at gRNA 5' terminus | ΔG > -1 kcal/mol improves efficiency | Facilitates R-loop formation and strand displacement |
| Poly-T/TTTT Motifs | Absence | Premature transcription termination if present | Acts as an RNA polymerase III terminator in U6-driven systems |
Chromatin Accessibility
The local epigenetic state is a major determinant of Cas9 binding and cutting.
Table 2: Epigenetic Features Correlating with gRNA Efficiency
| Feature | Assay/Marker | Correlation with Efficiency | Recommendation |
|---|---|---|---|
| DNase I Hypersensitivity | DNase-seq | Strong positive (R up to ~0.5) | Prioritize regions with high DHS signal |
| Histone Marks | H3K4me3, H3K9ac, H3K27ac (Active) | Positive correlation | Favor regions marked as transcriptionally active |
| DNA Methylation | CpG Methylation (e.g., WGBS) | Strong negative correlation for high methylation | Avoid densely methylated CpG islands near PAM |
Experimental Protocol: Validating gRNA On-Target Efficiency
This protocol outlines a method for empirical validation of gRNA cutting efficiency using next-generation sequencing (NGS) of PCR-amplified target sites.
Materials:
- Cell line of interest expressing Cas9 (stable or transient)
- gRNA expression vector (e.g., lentiGuide, pX459) or synthetic gRNA/Cas9 RNP
- Transfection or transduction reagents
- Genomic DNA extraction kit
- High-fidelity PCR master mix
- NGS library preparation kit compatible with amplicons
- Bioanalyzer/TapeStation for quality control
Procedure:
- Design & Cloning: Design 3-5 gRNAs per target locus using prediction tools (see Section 5). Clone oligos into your gRNA expression vector.
- Delivery: Deliver individual gRNA constructs into Cas9-expressing cells. Include a non-targeting control gRNA.
- Harvest Genomic DNA: 72-96 hours post-delivery, harvest cells and extract high-quality genomic DNA.
- Amplify Target Locus: Design primers ~150-300bp flanking each target site. Perform PCR with high-fidelity polymerase.
- NGS Library Prep & Sequencing: Purify PCR products, add sequencing adapters via a limited-cycle PCR, and pool for sequencing on an Illumina MiSeq or HiSeq platform (aim for >10,000x read depth per amplicon).
- Data Analysis: Use computational tools (e.g., CRISPResso2, ICE analysis) to align reads and quantify the percentage of insertions/deletions (indels) at the target site. Efficiency is calculated as (1 - % of unmodified reads).
Signaling Pathways in DNA Damage Response to Cas9 Cleavage
Cas9-induced double-strand breaks (DSBs) trigger a coordinated cellular DNA Damage Response (DDR), which influences editing outcomes and screen phenotypes.
Title: DNA Damage Response to Cas9-Induced Double-Strand Breaks
Predictive Models and Workflow for Optimal gRNA Selection
Modern gRNA selection integrates multiple sequence and epigenetic features into predictive algorithms.
Title: Integrated Workflow for Optimal gRNA Selection
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents for gRNA Design and Validation Experiments
| Item Category | Specific Example(s) | Function & Rationale |
|---|---|---|
| gRNA Expression Vector | lentiGuide-Puro, pSpCas9(BB)-2A-Puro (PX459) | Drives gRNA transcription from a U6 promoter; often includes a selection marker (e.g., puromycin). |
| Cas9 Cell Line | HEK293T-Cas9, HeLa-Cas9, or custom stable lines | Provides constitutive Cas9 expression, standardizing the nuclease component across screens. |
| Nuclease Delivery Reagent | Lipofectamine 3000, PEI Max (transfection); Lentiviral particles (transduction) | Enables efficient introduction of gRNA constructs into target cells. |
| gRNA Synthesis Reagent | Custom oligos for cloning; Synthetic sgRNA (e.g., from Trilink) | Source of the gRNA sequence. Synthetic sgRNA allows for rapid RNP complex delivery. |
| Genomic DNA Isolation Kit | DNeasy Blood & Tissue Kit (Qiagen), Quick-DNA Miniprep Kit (Zymo) | High-quality, PCR-ready genomic DNA is essential for accurate amplicon sequencing. |
| High-Fidelity PCR Mix | Q5 Hot Start (NEB), KAPA HiFi HotStart ReadyMix | Minimizes PCR errors during amplification of the target locus for NGS validation. |
| NGS Amplicon Library Prep Kit | Illumina DNA Prep, NEBNext Ultra II FS DNA Library Prep | Prepares barcoded sequencing libraries from PCR amplicons for multiplexed analysis. |
| Validation Analysis Software | CRISPResso2, ICE (Inference of CRISPR Edits) | Aligns NGS reads to reference and quantifies indel frequencies to measure cutting efficiency. |
Optimizing gRNA design is a non-trivial but essential step in CRISPR-Cas9 knockout screen research. A multi-factorial approach that combines thermodynamic sequence rules, chromatin context awareness, and empirical validation is necessary to predict and achieve high on-target efficiency. Integrating these principles into the screen design phase dramatically improves the reliability and interpretability of functional genomics data, accelerating discoveries in basic biology and drug development.
In the context of CRISPR-Cas9 knockout screen principle research, ensuring library representation is a foundational requirement for data integrity and biological discovery. A loss of representation—where specific single-guide RNAs (sgRNAs) or entire genes are underrepresented or lost from a pooled library during amplification, transduction, or screening—introduces severe biases, false negatives, and compromises statistical power. This technical guide details the calculations, monitoring protocols, and mitigation strategies essential for maintaining sufficient representation throughout a genome-wide or focused screen workflow, from library design to hit identification.
Core Principles: Calculating Representation and Coverage
The core metric for library quality is coverage, defined as the number of cells per sgRNA at the time of transduction. Sufficient coverage minimizes stochastic loss of sgRNAs due to random sampling.
Key Quantitative Parameters:
- N: The number of transduced cells.
- G: The number of sgRNAs in the library.
- MOI: Multiplicity of Infection (average number of viral integrations per cell). Target MOI < 0.3-0.4 to minimize multiple integrations per cell.
- Coverage (C): C = (N * MOI) / G
- Representation Threshold: A minimum coverage of 200-500x is standard for genome-wide screens. For essential gene identification or high-resolution phenotyping, ≥500-1000x coverage may be required.
Table 1: Coverage Calculation Examples for Common Library Scales
| Library Size (sgRNAs) | Target MOI | Transduced Cells Required for 200x Coverage | Transduced Cells Required for 500x Coverage |
|---|---|---|---|
| 10,000 (Focused) | 0.3 | ~6.67 million | ~16.67 million |
| 70,000 (Genome-wide) | 0.3 | ~46.67 million | ~116.67 million |
| 100,000 (Genome-wide) | 0.3 | ~66.67 million | ~166.67 million |
Calculation: Transduced Cells = (Coverage * Library Size) / MOI
Monitoring Representation: Experimental Protocols
Protocol: Pre-Screen Library Amplification & Quality Control
Objective: Generate sufficient plasmid and viral library complexity without skewing.
- Transformation: Use electrocompetent cells with high transformation efficiency (>1e9 cfu/µg). Use at least 1000x the library size in colony-forming units (e.g., 100 million colonies for a 100k sgRNA library).
- Plasmid Harvest: Grow transformed bacteria in large, liquid culture (≥1L), ensuring the total number of cells greatly exceeds the library size to maintain representation. Use maxiprep or megaprep kits designed for high-yield, low-shear DNA purification.
- NGS Validation (Plasmid Library):
- Amplify: PCR amplify the sgRNA cassette from 100-200ng of plasmid prep using indexing primers for Illumina sequencing.
- Sequence: Perform shallow sequencing (∼50-100 reads per sgRNA).
- Analyze: Calculate the read count per sgRNA. A high-quality library will show a tight, unimodal distribution of log-normalized reads. >99% of sgRNAs should be within 100-fold of the median read count.
Table 2: QC Metrics for Plasmid and Viral Libraries
| QC Step | Metric | Acceptance Criterion |
|---|---|---|
| Plasmid Library | sgRNAs Detected by NGS | >99.5% of expected sgRNAs |
| Plasmid Library | Read Distribution | Even log-normal distribution; no extreme outliers |
| Viral Titer | Functional Titer (TU/mL) | Accurately determined via puromycin selection or GFP |
| Viral Library | Infection Efficiency | Matches expectation for cell line (e.g., 30-60%) |
| Viral Library | sgRNA Representation (Post-Transduction) | Strong correlation with plasmid library (R² > 0.9) |
Protocol: Assessing Representation at Cell Transduction
Objective: Verify maintenance of library complexity post-transduction and pre-selection.
- Harvest Genomic DNA (gDNA): 48-72 hours post-transduction (pre-selection), harvest a pilot sample of cells (∼5-10 million). Extract high-molecular-weight gDNA.
- Amplify sgRNA Cassettes: Perform a first-round PCR from 1-2µg of gDNA to amplify integrated sgRNA sequences. Use a limited number of PCR cycles (∼10-12) to avoid skewing.
- Index for Sequencing: Perform a second-round, limited-cycle PCR to add Illumina adapters and sample indices.
- Sequencing & Analysis: Perform shallow sequencing. Compare sgRNA abundance distribution to the original plasmid library. A strong Spearman correlation (ρ > 0.98) indicates maintained representation.
Maintaining Representation During Screen Execution
Critical Steps and Mitigations:
- Cell Culture: Maintain cells in exponential growth. Never let cultures overgrow. Use population sizes that are always >> (Library Size * Desired Coverage) at all passages.
- Harvesting & Splitting: Use gentle centrifugation and thorough resuspension to avoid clumping. Always harvest a random sample of the entire population.
- gDNA Harvest for Timepoints: For the final T0 (post-selection) and T-end samples, harvest gDNA from a number of cells that guarantees maintained coverage. A rule of thumb is to harvest 1000x library size in cells (e.g., 100 million cells for a 100k library).
- PCR Amplification for Deep Sequencing: Distribute gDNA across multiple independent PCR reactions (≥4-8 reactions per sample) to minimize PCR bias. Pool reactions after cleanup.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Library Representation QC
| Item | Function & Critical Feature |
|---|---|
| High-Efficiency Electrocompetent Cells (e.g., Endura, Stbl4) | Ensures transformation with complexity >1000x library size; reduces recombination of repetitive sgRNA vectors. |
| Large-Scale Plasmid Prep Kit (e.g., Maxi/Mega/Giga Prep) | High-yield, high-purity DNA prep for viral production without mechanical shearing. |
| Next-Generation Sequencing Kit (Illumina-compatible) | For quantifying sgRNA abundance in plasmid, viral, and genomic DNA libraries. |
| High-Fidelity, Low-Bias PCR Polymerase (e.g., KAPA HiFi, Herculase II) | Critical for unbiased amplification of sgRNA cassettes from gDNA for NGS library prep. |
| Genomic DNA Extraction Kit (Scalable, Spin-Column or Liquid Handling) | For clean gDNA isolation from 1 million to 1 billion cells. Must minimize shearing. |
| Lentiviral Packaging Mix (3rd Gen.) | For producing high-titer, replication-incompetent lentiviral sgRNA library. |
| Polybrene or Hexadimethrine Bromide | Enhances viral transduction efficiency in hard-to-transduce cells. |
| Puromycin or Appropriate Selection Antibiotic | For selecting successfully transduced cells post-viral infection. |
| Cell Counter (Automated) | For accurate determination of cell numbers at transduction and during passaging to maintain coverage. |
| Flow Cytometer | For precise determination of viral transduction efficiency (if using a fluorescent marker). |
Visualizing Workflows and Relationships
Title: CRISPR Screen Workflow with QC Checkpoints
Title: Coverage Calculation Logic Loop
In CRISPR-Cas9 knockout screening, false positives (genes identified as hits that are not biologically relevant) and false negatives (true hits missed by the screen) directly compromise the validity of functional genomics studies and downstream drug target identification. This guide details the systematic experimental and analytical framework required to mitigate these errors, ensuring robust, reproducible results for therapeutic discovery.
False Positives: Arise from off-target CRISPR effects, genetic or phenotypic heterogeneity, assay technical noise, and batch effects. False Negatives: Result from incomplete gene knockout, low sgRNA activity, low sequencing depth, and suboptimal assay sensitivity.
The triad of Controls, Replicates, and Analytical Thresholds forms the foundational strategy for error mitigation.
Essential Controls for Screen Validation
Table: Critical Control Types in CRISPR Screens
| Control Type | Purpose | Recommended Implementation | Mitigates |
|---|---|---|---|
| Non-Targeting Controls (NTCs) | Define baseline signal and null distribution. | 50-1000 sgRNAs with no homology to the genome. Scatter throughout library. | False Positives (assay noise) |
| Positive Controls | Assess screen dynamic range and sgRNA activity. | Essential genes (e.g., ribosomal, proteasome) expected to drop out in viability screens. | False Negatives (technical failure) |
| Seed Controls | Control for sequence-specific, microRNA-like off-target effects. | sgRNAs with matching "seed" region but different PAM/distal sequence. | False Positives (off-target) |
| Copy-Number Controls | Account for proliferation effects due to copy number alterations. | Target genomic regions with neutral copy number in cell model. | False Positives (CNV effects) |
| Treatment Controls | Isolate effect of selection agent from genetic perturbation. | Cells transduced with library but not subjected to selection pressure. | False Positives (selection bias) |
Detailed Protocol: Designing and Implementing Non-Targeting Controls
- Design: Generate 500-1000 20nt sequences with no significant homology (≤12 bp contiguous match) to the target genome using algorithms like
BowtieorBLAST. Ensure identical length and GC content distribution as targeting sgRNAs. - Cloning: Synthesize oligonucleotides and clone them into the chosen sgRNA backbone (e.g., lentiCRISPR v2, pXPR vectors) using BsmBI restriction sites via Golden Gate assembly.
- Integration: Mix NTCs uniformly with the targeting sgRNA library. Co-package into lentivirus at a low MOI (<0.3) to ensure single integrations.
- Application: Use NTC read counts across all samples to:
- Normalize read counts (e.g., median-of-ratios).
- Model the null distribution for statistical testing (e.g., using
MAGeCKorCRISPRcleanR). - Set empirical false discovery rate (FDR) thresholds.
The Role of Replicates: Biological vs. Technical
Table: Replicate Strategy for Robust Screening
| Replicate Type | Definition | Primary Purpose | Minimum Recommended Number |
|---|---|---|---|
| Technical Replicate | Multiple sequencing runs or PCR amplifications of the same biological sample. | Quantify and reduce sequencing/PCR noise. | 2 (for sequencing) |
| Biological Replicate | Independently transduced, selected, and processed cell populations from the same cell line/pool. | Account for stochastic variation in transduction, clonal heterogeneity, and library representation. | 3-4 |
| Experimental Replicate | Entire screen performed independently on different days/cell passages. | Capture broader technical variability and ensure reproducibility. | 2 |
Protocol: Performing Biological Replicates in a Pooled Screen
- Day 1: Seeding. Plate the same parental cell line into 3-4 independent culture vessels.
- Day 2: Transduction. For each replicate, transduce cells with the same lentiviral library prep but using separate culture vessels. Maintain identical MOI (~0.3) and cell numbers.
- Post-Transduction: Culture each replicate independently through antibiotic selection and the experimental timeline (e.g., drug treatment, time passaging).
- Harvest & Processing: Harvest genomic DNA from each replicate pellet separately. Perform independent PCR amplifications of the sgRNA region with unique barcoded primers for each replicate.
- Analysis: Sequence replicates separately. Use robust statistical models (e.g., in
MAGeCKorPinAPL-Py) that account for variance between replicates to call significant hits, increasing the degrees of freedom and statistical power.
Establishing Analytical Thresholds
| Analytical Parameter | Typical Range/Value | Calculation/Definition | Impact on Error | ||
|---|---|---|---|---|---|
| Minimum Read Depth | 200-500 reads per sgRNA | Total reads / (Library Size * Coverage). Lower depth increases FN. | Mitigates False Negatives | ||
| Fold-Change Cutoff | Varies (e.g., | LFC | > 0.5 - 1) | Log2(Treatment/Control). Too stringent increases FN; too lenient increases FP. | Balances FP/FN |
| Statistical Threshold | FDR < 0.05 - 0.25; p-value < 0.05 | Corrected for multiple hypothesis testing (Benjamini-Hochberg). Primary guard against FP. | Mitigates False Positives | ||
| sgRNA Consistency | ≥ 2/3 sgRNAs per gene agree | Number of sgRNAs for a gene showing same-direction significant effect. | Mitigates False Positives | ||
| Gene Essentiality Z-score | Z | > 2 | Robust Z-score based on negative control sgRNA distribution. | Mitigates False Positives |
Protocol: Determining the Optimal False Discovery Rate (FDR) Threshold
- Screen Analysis: Run primary analysis (read alignment, count normalization, LFC calculation) using a tool like
MAGeCKorCRISPRcleanR. - Null Distribution Modeling: The tool uses the NTCs to model the expected distribution of LFCs under the null hypothesis (no effect).
- p-value and FDR Calculation: For each gene, a p-value is computed (e.g., via robust rank aggregation of its sgRNAs). The FDR (q-value) is calculated using the Benjamini-Hochberg procedure across all genes.
- Threshold Selection: Plot the ranked gene list by q-value. A common threshold is FDR < 0.1. Stricter thresholds (FDR < 0.05) reduce FPs but may increase FNs. Validate the chosen threshold using the positive control genes—they should be highly significant.
Visualization of Workflows and Logic
Diagram 1: End-to-end screen workflow with replicates
Diagram 2: Hit-calling logic using sequential thresholds
The Scientist's Toolkit: Research Reagent Solutions
| Item/Category | Example Product/Supplier | Function in Mitigating Error |
|---|---|---|
| Validated sgRNA Library | Brunello, TKOv3 (Addgene), Human CRISPR Knockout (Horizon) | Pre-designed libraries with high on-target scores, included NTCs, and essential gene positive controls to reduce design-based FPs/FNs. |
| Lentiviral Packaging Mix | Lenti-X, psPAX2/pMD2.G (Takara, Addgene) | Produces high-titer, consistent virus for uniform transduction (MOI~0.3), minimizing variance that leads to FNs. |
| Next-Gen Sequencing Kit | Illumina NovaSeq, MiSeq Reagent Kits | Provides deep, uniform sequencing coverage (>500x per sgRNA) to accurately quantify abundance, reducing FNs from dropout. |
| gDNA Isolation Kit | Quick-DNA Midiprep Kit (Zymo Research) | High-yield, pure gDNA extraction from large cell pellets (≥ 1e7 cells) for reproducible PCR amplification of sgRNAs. |
| sgRNA Amplification Primers | Indexed P5/P7 Primers (IDT) | Unique dual-indexed primers for multiplexing biological replicates, allowing direct variance measurement and batch correction. |
| Cell Viability Assay | CellTiter-Glo (Promega) | Validates positive control dropout and screen dynamic range in viability screens, confirming assay sensitivity. |
| Analysis Software Suite | MAGeCK, PinAPL-Py, CRISPRcleanR | Implements robust statistical models using negative controls and replicates to calculate FDR and LFC, the core of threshold setting. |
| Essential Gene Reference | CRISPR Essentialome (DepMap) | Public dataset of common essential genes used as benchmark positive controls to calibrate screen performance and thresholds. |
CRISPR-Cas9 knockout screening has evolved from a fundamental tool for identifying gene function in vitro to a sophisticated platform for probing complex biological systems. This whitepaper details advanced applications that extend the principle of pooled genetic perturbation into more physiologically relevant and functionally nuanced domains. The core thesis of knockout screen research—correlating genetic loss-of-function with phenotypic readout—is now being applied within living organisms, expanded to dissect genetic interactions, and refined through reversible transcriptional modulation.
In Vivo CRISPR Screening
In vivo screening transplants the principles of pooled library screening from cell culture into animal models, typically mice. This allows for the identification of genes essential for processes like tumor growth, metastasis, immune evasion, and response to therapy within a complex tissue microenvironment.
Core Methodology & Protocol
Protocol: In Vivo Positive Selection Screening for Tumor Fitness Genes
- Library Transduction: Infect a population of tumor cells (e.g., mouse or human cancer cell line) with a lentiviral sgRNA library (e.g., Brunello or Brie genome-wide library) at a low Multiplicity of Infection (MOI ~0.3) to ensure most cells receive one sgRNA.
- Selection and Expansion: Select transduced cells with puromycin for 3-5 days. Expand the population for 7-10 days to allow for gene knockout.
- Baseline Sample (T0): Harvest at least 50 million cells (providing ~1000x coverage of the library) and extract genomic DNA (gDNA).
- Injection and In Vivo Growth: Inject 5-10 million library-transduced cells subcutaneously or orthotopically into immunocompromised (e.g., NSG) or immunocompetent syngeneic mice. Use sufficient mice to maintain >500x library coverage.
- Endpoint Sample (T1): After tumor growth (e.g., 4-8 weeks), harvest tumors, dissociate into single cells, and extract gDNA.
- NGS Library Prep & Analysis: Amplify integrated sgRNA sequences from gDNA via PCR, sequence, and quantify sgRNA abundance. Compare T1/T0 abundance using MAGeCK or BAGEL2 algorithms to identify significantly depleted or enriched sgRNAs.
Table 1: Key Considerations for In Vivo Screen Design
| Parameter | Typical Specification | Rationale |
|---|---|---|
| Library Size | 4-10 sgRNAs/gene | Balances depth with practical animal numbers. |
| Cell Coverage | >500x per sample | Ensures statistical power to detect dropout. |
| Mouse Cohort | 3-5 mice per group/condition | Accounts for inter-animal variability. |
| Tumor Harvest | At defined volume (e.g., 1000 mm³) or timepoint | Standardizes selective pressure. |
The Scientist's Toolkit: In Vivo Screening
| Research Reagent Solution | Function |
|---|---|
| Focused sgRNA Library (e.g., Metabolic, Kinase, Tumor Suppressor) | Reduces library size for higher in vivo coverage; targets biologically relevant gene sets. |
| Barcoded Lentiviral Vectors | Allows multiplexing of different cell lines or conditions in the same animal (CellTagging). |
| Next-Gen Sequencing Kit (e.g., Illumina MiSeq) | For high-throughput sgRNA quantification from tumor-derived gDNA. |
| Single-Cell RNA-Seq Solutions | Enables coupling of genetic perturbation with transcriptional profiling in vivo (CRISPR-sci). |
| Immunocompromised Mouse Strains (NSG, NOG) | Supports engraftment of human xenografts for screens in a humanized context. |
Title: Workflow for In Vivo CRISPR Knockout Screening
Combinatorial Genetic Knockouts
Combinatorial knockout screening aims to identify genetic interactions—synthetic lethality or synergy—by targeting two or more genes simultaneously within a single cell. This reveals functional redundancies and pathway cross-talk.
Experimental Protocol: Dual-Knockout Screening with Paired sgRNAs
Protocol: Arrayed Dual-gRNA Virus Production & Screening
- Library Design: Create an arrayed library in a 96- or 384-well format where each well contains a pair of sgRNAs targeting two distinct genes (or non-targeting controls).
- Virus Production: In each well of a culture plate, co-transfect HEK293T cells with three plasmids: a lentiviral backbone containing sgRNA pair, psPAX2 (packaging), and pMD2.G (envelope). Use PEI or calcium phosphate transfection.
- Viral Harvest: Collect lentiviral supernatant at 48 and 72 hours post-transfection, filter (0.45 µm), and optionally concentrate.
- Cell Infection: Infect target cells (seeded in assay plates) with the arrayed virus in the presence of polybrene (8 µg/mL). Spinfect at 1000 x g for 30-60 minutes.
- Phenotypic Assay: After 5-10 days for gene knockout, assay each well for phenotype (e.g., cell viability via ATP-based luminescence, imaging).
- Analysis: Normalize luminescence to controls. Calculate combinatorial scores (e.g., Bliss Independence score) to classify interactions as synthetic lethal, additive, or antagonistic.
Table 2: Metrics for Analyzing Genetic Interactions
| Interaction Type | Mathematical Definition (Bliss) | Interpretation |
|---|---|---|
| Synthetic Lethality/Sickness | Observed Effect < (EffectA + EffectB - EffectA*EffectB) | Combined knockout is more deleterious than expected. |
| Additive | Observed Effect ≈ (EffectA + EffectB - EffectA*EffectB) | Combined effect equals the sum of individual effects. |
| Antagonistic/Suppressive | Observed Effect > (EffectA + EffectB - EffectA*EffectB) | Combined knockout is less deleterious than expected. |
Title: Combinatorial Knockout Screen for Genetic Interactions
CRISPR Interference and Activation (CRISPRi/a) Integration
CRISPRi (interference) and CRISPRa (activation) utilize a catalytically dead Cas9 (dCas9) fused to transcriptional repressor (e.g., KRAB) or activator (e.g., VP64-p65-Rta) domains. This allows for reversible, sequence-specific gene knockdown or overexpression without altering the genomic DNA, enabling gain- and loss-of-function screens.
Key Protocols
Protocol A: Stable Cell Line Generation for CRISPRi/a
- dCas9 Effector Line Creation: Lentivirally transduce target cells with dCas9-KRAB (for i) or dCas9-VPR (for a). Select with blasticidin (common resistance marker) for 10-14 days.
- Validation: Test functionality by transducing with sgRNAs targeting a known essential gene (for i) or a silent reporter gene (for a) and measuring phenotype. Protocol B: CRISPRi/a Pooled Screening
- Library Transduction: Infect the stable dCas9-expressing cell line with a genome-wide sgRNA library (targeting transcription start sites for CRISPRa, or gene bodies for CRISPRi) at low MOI.
- Selection & Phenotype Application: Select with puromycin (on the sgRNA vector). Apply a phenotypic selection (e.g., drug treatment, nutrient deprivation) or simply passage cells for fitness screens.
- Analysis: Harvest gDNA at T0 and T1, sequence sgRNAs, and analyze similarly to knockout screens.
Table 3: Comparison of CRISPR Knockout, Interference, and Activation
| Feature | CRISPR Knockout | CRISPR Interference (i) | CRISPR Activation (a) |
|---|---|---|---|
| Cas9 Form | Wild-type (Nuclease) | dCas9-Repressor (e.g., KRAB) | dCas9-Activator (e.g., VPR) |
| Genetic Change | Permanent indel mutation | Epigenetic, reversible | Epigenetic, reversible |
| Effect on Gene | Complete, permanent loss | Transcriptional knockdown (up to ~90%) | Transcriptional overexpression (up to 100x) |
| Screen Application | Essential genes, fitness | Hypomorphic phenotypes, essential gene studies | Gain-of-function, drug resistance, differentiation |
| Key Target Site | Early exons | TSS (-50 to +300 bp) | TSS (-50 to +300 bp) or enhancer regions |
The Scientist's Toolkit: CRISPRi/a
| Research Reagent Solution | Function |
|---|---|
| dCas9-KRAB Lentiviral Construct | Stable expression of the CRISPR interference effector protein. |
| dCas9-VPR Lentiviral Construct | Stable expression of the CRISPR activation effector protein. |
| CRISPRi/a-Optimized sgRNA Libraries | Libraries designed with sgRNAs targeting transcriptional start sites (TSS). |
| Blasticidin & Puromycin | Antibiotics for selecting dCas9 effector cells and sgRNA-containing cells, respectively. |
| RT-qPCR Kits | For rapid validation of gene knockdown or activation efficiency prior to screening. |
Title: Core Mechanism of CRISPR Interference and Activation
Integrated Workflow and Concluding Outlook
The convergence of these advanced applications represents the next frontier in functional genomics. A modern, integrated screening pipeline may involve using CRISPRi/a for primary hit identification in vitro, followed by validation with combinatorial knockouts, and final confirmation in an in vivo model. The consistent underlying principle remains the correlation of a directed genetic perturbation with a high-dimensional phenotypic readout, now scalable to the complexity of living systems and the interactome.
Validating Hits and Choosing Your Tool: CRISPRko vs. Alternative Functional Genomic Methods
CRISPR-Cas9 knockout screens have revolutionized functional genomics, enabling genome-wide identification of genes essential for specific biological processes, such as cell viability, drug resistance, or pathway activation. The core thesis of this principle research is that systematic gene knockout, followed by selective pressure, reveals genetic dependencies. However, primary screening data is inherently noisy, containing both false positives (e.g., off-target effects, variable sgRNA efficiency) and false negatives. Therefore, the critical step in translating screen findings into credible biological insights or drug targets is the rigorous validation of candidate hits through orthogonal, secondary assays. This guide details the rationale, methodologies, and tools for this essential validation phase.
Primary Screen Hit Categorization & Validation Rationale
Primary screens generate quantitative data, typically analyzed via next-generation sequencing of sgRNA abundance. Key metrics for hit identification are summarized below.
Table 1: Common Metrics for Identifying Hits in CRISPR Knockout Screens
| Metric | Calculation | Hit Threshold | Interpretation |
|---|---|---|---|
| Log2 Fold Change (LFC) | log2(Post-selection sgRNA count / Initial sgRNA count) | LFC < -1 (dropout) or >1 (enrichment) | Magnitude of phenotype strength. |
| p-value | Statistical significance of sgRNA depletion/enrichment vs. control (e.g., MAGeCK, DESeq2). | p < 0.05 | Likelihood the effect is not due to chance. |
| False Discovery Rate (FDR) | Corrected p-value (e.g., Benjamini-Hochberg). | FDR < 0.25 (common in screens) or <0.1 | Estimated proportion of false positives among hits. |
| Gene Robustness Rank | Consistency of phenotype across multiple targeting sgRNAs. | Top 10% of ranked genes | Confirms on-target effect. |
Hits from Table 1 require validation to rule out artifacts and confirm the genotype-phenotype link.
Secondary Assay Methodologies
Orthogonal Genetic Validation
This confirms the phenotype is due to knockout of the specific gene.
Protocol A: CRISPR-Cas9 Mediated Knockout with Independent sgRNAs
- Objective: Reproduce phenotype using new sgRNAs targeting different exons of the hit gene.
- Steps:
- Design 2-3 new sgRNAs (using tools like CHOPCHOP or Benchling).
- Clone into lentiviral sgRNA expression vector (e.g., lentiGuide-Puro).
- Transduce into Cas9-expressing target cells. Include non-targeting sgRNA control.
- Select with puromycin (1-3 µg/mL, 48-72 hours).
- After selection, assay phenotype (e.g., proliferation, drug sensitivity) 5-7 days post-transduction.
- Confirm knockout efficiency via western blot (if antibody available) or T7 Endonuclease I assay / Tracking of Indels by Decomposition (TIDE) analysis on genomic DNA.
Protocol B: RNA Interference (RNAi) Knockdown
- Objective: Orthogonal validation using a different loss-of-function mechanism.
- Steps:
- Obtain 2-3 independent siRNA or shRNA sequences targeting the hit gene.
- Transfert siRNA (lipofection/electroporation) or transduce shRNA lentivirus into wild-type cells.
- Assay phenotype 72-96 hours (siRNA) or after stable selection (shRNA).
- Confirm mRNA knockdown via qRT-PCR.
Phenotypic Validation in Relevant Models
Protocol C: Cell Titer-Glo Viability Assay
- Objective: Quantitatively measure proliferation/viability impact.
- Steps:
- Seed validated knockout and control cells in 96-well plates (500-2000 cells/well).
- Incubate for desired time course (e.g., 1-5 days).
- Equilibrate plate to room temperature for 30 minutes.
- Add equal volume of Cell Titer-Glo reagent, mix for 2 minutes, incubate in dark for 10 minutes.
- Record luminescence. Plot relative luminescence vs. time.
Protocol D: Competitive Co-culture Assay by Flow Cytometry
- Objective: Precisely measure fitness defects in a mixed population.
- Steps:
- Generate knockout cells expressing a fluorescent marker (e.g., GFP).
- Mix them at a 1:1 ratio with control (RFP-expressing) cells.
- Culture the mix over 7-14 days, sampling periodically.
- Analyze GFP+/RFP+ ratio by flow cytometry.
- Calculate relative fitness: s = ln[(GFPt/RFPt) / (GFP0/RFP0)] / t.
Visualizing Validation Workflows & Pathways
Workflow for Validating CRISPR Screen Hits
Mechanistic Insight from a Validated Hit
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for CRISPR Hit Validation
| Item | Function & Application | Example Products/Tools |
|---|---|---|
| Lentiviral sgRNA Vectors | Deliver validation sgRNAs; enable stable selection. | lentiGuide-Puro (Addgene #52963), pKLV2 (Sigma). |
| Cas9-Expressing Cell Lines | Provide constant Cas9 for knockout with sgRNA alone. | Commercially available lines or generate via lentivirus (lentiCas9-Blast). |
| siRNA/shRNA Libraries | For orthogonal RNAi knockdown. | Dharmacon ON-TARGETplus siRNA, TRC shRNA clones. |
| Cell Viability Assay Kits | Quantify phenotypic impact of knockout. | Cell Titer-Glo 3D (Promega), MTT/WST-8 assays. |
| Genomic DNA Extraction Kits | Isolate DNA for knockout efficiency analysis. | QuickExtract (Lucigen), DNeasy (Qiagen). |
| Knockout Verification Tools | Assess indel formation at target locus. | TIDE web tool, T7 Endonuclease I (NEB), ICE (Synthego). |
| Antibodies for Western Blot | Confirm protein-level knockout (gold standard). | Validate via resources like Antibodypedia or vendor data. |
| Flow Cytometry Markers | Enable competitive co-culture assays. | Lentiviral GFP/RFP constructs, cell tracking dyes. |
| NGS Library Prep Kits | Validate sgRNA representation if performing pooled validation. | Nextera XT (Illumina), SMARTer smRNA-Seq (Takara). |
This in-depth technical guide provides a comparative analysis of two foundational techniques in CRISPR-Cas9-based genetic screening: CRISPR Knockout (CRISPRko) and CRISPR Interference/Activation (CRISPRi/a). Framed within the broader thesis of CRISPR Cas9 knockout screen principle research, this document serves as a critical resource for selecting the optimal perturbation method for functional genomics studies and drug target discovery. CRISPRko utilizes the endonuclease activity of Cas9 to create double-strand breaks (DSBs), leading to frameshift mutations and gene disruption via non-homologous end joining (NHEJ). In contrast, CRISPRi/a employs a catalytically "dead" Cas9 (dCas9) fused to effector domains to repress (i/a) or activate (a) gene transcription without altering the underlying DNA sequence. The choice between these systems hinges on experimental goals, including the desired perturbation type (permanent vs. reversible), screening context (essential gene identification vs. subtle phenotypic analysis), and biological question.
Core Mechanisms & Molecular Biology
CRISPR Knockout (CRISPRko)
CRISPRko relies on the wild-type Streptococcus pyogenes Cas9 (SpCas9) nuclease. A single-guide RNA (sgRNA) directs Cas9 to a complementary genomic locus adjacent to a Protospacer Adjacent Motif (PAM; NGG for SpCas9). Cas9 generates a blunt-ended DSB 3 bp upstream of the PAM. In mammalian cells, the dominant repair pathway, NHEJ, frequently introduces small insertions or deletions (indels) at the break site. When these indels occur within a protein-coding exon, they can cause frameshifts and premature stop codons, resulting in a loss-of-function allele.
CRISPR Interference (CRISPRi)
CRISPRi uses a nuclease-deficient dCas9 (carrying D10A and H840A mutations) that binds DNA but does not cleave it. For repression, dCas9 is fused to a transcriptional repressor domain, such as the Krüppel-associated box (KRAB) from human Kox1. When targeted to a transcription start site (TSS) or promoter region, the dCas9-KRAB fusion protein recruits heterochromatin-forming complexes, leading to histone methylation (H3K9me3) and subsequent gene silencing. Effective silencing typically requires targeting within -50 to +300 bp relative to the TSS.
CRISPR Activation (CRISPRa)
CRISPRa also utilizes dCas9 but is fused to transcriptional activator domains. Common systems include dCas9-VP64 (a tetramer of the Herpes Simplex Viral Protein 16), which is often combined with additional RNA scaffolds (e.g., MS2, PP7) that recruit further activator proteins (e.g., p65, HSF1) to form a "synergistic activation mediator" (SAM) complex. Targeting is typically within -400 to -50 bp upstream of the TSS to recruit the cellular transcription machinery and upregulate gene expression.
Diagram Title: Core Mechanisms of CRISPRko, i, and a
Quantitative Comparison & Performance Metrics
The following tables summarize key performance characteristics of each technology, based on recent literature and benchmarking studies.
Table 1: Fundamental Operational Parameters
| Parameter | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Cas9 Variant | Wild-type SpCas9 (Nuclease) | dCas9 (D10A, H840A) | dCas9 (D10A, H840A) |
| Core Effector | Nuclease Domain | Repressor Domain (e.g., KRAB) | Activator Domain (e.g., VP64, SAM) |
| DNA Cleavage | Yes (DSB) | No | No |
| Genomic Change | Permanent (Indels) | Epigenetic/None | Epigenetic/None |
| Perturbation Type | Loss-of-function (knockout) | Loss-of-function (knockdown) | Gain-of-function (overexpression) |
| Typical On-Target Efficacy | >80% frameshift rate (highly active sgRNAs) | 70-95% knockdown (protein level) | 5-50x mRNA upregulation (varies by gene) |
| Reversibility | Irreversible | Reversible (upon dCas9 depletion) | Reversible (upon dCas9 depletion) |
| Key Targeting Region | Early exons (coding sequence) | -50 to +300 bp from TSS | -400 to -50 bp from TSS |
Table 2: Performance in Genome-Wide Screens
| Metric | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Library Size (Human) | ~90,000 sgRNAs (3-4/gene) | ~110,000 sgRNAs (5-10/gene) | ~70,000 sgRNAs (5-10/gene) |
| Optimal Screen Readout | Cell proliferation/survival (essential genes), resistance/sensitivity | Sensitive phenotypes (e.g., differentiation, subtle fitness), synthetic lethality | Gain-of-function phenotypes (e.g., drug resistance, oncogene activation) |
| False Positive Rate | Low (but can have false positives from DSB toxicity/p53 response) | Very Low (minimal DNA damage) | Low (potential for off-target activation) |
| False Negative Rate | Moderate (ineffective sgRNAs, redundancy) | Low-Moderate (position-dependent efficacy) | Moderate-High (highly context-dependent activation) |
| Typical Hit Concordance (vs. RNAi) | High for core essentials | Higher specificity, fewer off-targets than RNAi | N/A (complementary approach) |
| Time to Phenotype | Days to weeks (requires protein turnover) | Hours to days (rapid transcriptional effect) | Hours to days (rapid transcriptional effect) |
Detailed Experimental Protocols
Protocol for CRISPRko Negative Selection Screen
Objective: To identify genes essential for cell proliferation/survival under standard culture conditions.
Materials & Workflow:
- Library Design & Cloning: Use a validated genome-wide lentiviral sgRNA library (e.g., Brunello, Brie). Clone pool into lentiviral transfer plasmid with puromycin resistance.
- Virus Production: Co-transfect HEK293T cells with the library plasmid, psPAX2 (packaging), and pMD2.G (VSV-G envelope) using PEI transfection reagent. Harvest supernatant at 48h and 72h, concentrate via ultracentrifugation.
- Cell Transduction & Selection: Titrate virus on target cells. Transduce cells at an MOI of ~0.3 to ensure most cells receive a single sgRNA. Maintain a minimum of 500x library coverage. Select with puromycin (1-2 µg/mL) for 5-7 days.
- Screen Passage & Harvest: Passage cells every 2-3 days, maintaining >500x coverage. Harvest genomic DNA from ~50 million cells at the initial time point (T0) and after 14-21 population doublings (Tfinal) using a maxiprep kit.
- Amplification & Sequencing: Amplify integrated sgRNA cassettes from gDNA via two-step PCR (Primers: add Illumina adapters and sample barcodes). Purify PCR products and sequence on an Illumina NextSeq platform (75bp single-end).
- Data Analysis: Align reads to the reference sgRNA library. Count sgRNA reads for T0 and Tfinal. Normalize counts, calculate log2 fold-change for each sgRNA. Use a model (e.g., MAGeCK, BAGEL) to rank essential genes based on sgRNA depletion.
Diagram Title: CRISPRko Negative Selection Screen Workflow
Protocol for CRISPRi/a Positive Selection Screen
Objective (CRISPRa): To identify genes whose overexpression confers resistance to a targeted anticancer drug.
Materials & Workflow:
- Library Design & Cell Line Engineering: Use a targeted CRISPRa sgRNA library (e.g., Calabrese library) focusing on kinase/TF genes. First, generate a stable cell line expressing the dCas9-activator (e.g., SAM system) via lentiviral transduction and blasticidin selection.
- Screen Transduction & Selection: Transduce the engineered cell line with the sgRNA library at MOI~0.3. Select with puromycin for 5 days.
- Drug Challenge: Split cells into vehicle (DMSO) and drug-treated arms. Treat with the IC90 concentration of the drug. Passage cells, maintaining >500x coverage for 14-21 days.
- Harvest & Sequencing: Harvest gDNA from pre-selection (T0), vehicle, and drug-treated (Tfinal) populations. Amplify and sequence sgRNA inserts as in Protocol 4.1.
- Data Analysis: Compare sgRNA abundance between drug-treated and vehicle control populations. Enriched sgRNAs indicate genes whose activation promotes drug resistance. Use MAGeCK or similar tool for statistical analysis.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for CRISPR Functional Screens
| Item (Example Product) | Function | Key Consideration |
|---|---|---|
| Genome-wide sgRNA Library (Brunello ko, Dolcetto i/a) | Pre-designed, pooled sgRNA sets for targeting every gene. | Optimized for on-target efficiency and reduced off-target effects. Delivered as arrayed oligonucleotides or cloned plasmid pools. |
| Lentiviral Transfer Plasmid (lentiCRISPRv2, lentiGuide-Puro) | Backbone for sgRNA expression, includes selection marker (e.g., PuroR). | May contain Cas9 (for ko) or require separate dCas9-effector line (for i/a). |
| dCas9-Effector Plasmid (pHAGE-dCas9-KRAB, lenti SAMv2) | For stable expression of dCas9 fused to repressor (KRAB) or activator (SAM). | Required for CRISPRi/a. Must be stably expressed before sgRNA transduction. |
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Third-generation system for producing replication-incompetent lentivirus. | Essential for safe and efficient delivery of CRISPR components. |
| Polyethylenimine (PEI) Transfection Reagent | For co-transfection of plasmids into HEK293T cells to produce virus. | Cost-effective, high-efficiency alternative to commercial lipid reagents. |
| Selection Antibiotics (Puromycin, Blasticidin) | To select for cells successfully transduced with CRISPR constructs. | Titrate kill curve for each cell line; use minimal effective concentration. |
| gDNA Extraction Kit (Maxi/Midi Prep, e.g., Qiagen) | To harvest high-quality, high-quantity genomic DNA from pooled cell populations. | Scalability and yield are critical for maintaining library representation. |
| High-Fidelity PCR Kit (e.g., KAPA HiFi) | For accurate, low-bias amplification of sgRNA sequences from gDNA. | Essential to prevent skewing of sgRNA abundance during NGS prep. |
| Illumina Sequencing Reagents | For high-throughput sequencing of sgRNA amplicons. | Single-end 75bp runs are typically sufficient. |
| Analysis Software (MAGeCK, BAGEL, CRISPResso2) | For quantifying sgRNA depletion/enrichment and identifying hit genes. | MAGeCK is the current standard for robust statistical analysis. |
CRISPRko and CRISPRi/a are complementary technologies that address distinct biological questions within the framework of CRISPR screen principle research. CRISPRko is the gold standard for identifying essential genes and creating permanent, complete loss-of-function, making it ideal for synthetic lethality and robust survival screens. CRISPRi offers reversible, titratable knockdown with minimal off-target confounding from DNA damage, excelling in studies of sensitive phenotypes, non-coding genomic elements, and essential gene phenotyping where knockout is lethal. CRISPRa enables systematic gain-of-function screening, a unique capability for discovering genes that drive resistance, differentiation, or other activation-based phenotypes. The selection of the appropriate technology hinges on the specific research thesis, with considerations for the nature of the desired genetic perturbation, phenotypic sensitivity, and the required experimental timeline. Future developments in Cas orthologs, effector domains, and screening modalities will continue to expand the precision and scope of these foundational tools.
Within the broader thesis of CRISPR-Cas9 knockout screen principle research, understanding the comparative landscape of functional genomic screening technologies is fundamental. For over a decade, RNA interference (RNAi) was the dominant technique for loss-of-function screens. The advent of CRISPR-Cas9-mediated knockout has revolutionized the field, offering distinct advantages and revealing limitations when contrasted with its predecessor. This technical guide provides an in-depth comparison of these two pivotal technologies, focusing on their mechanisms, experimental protocols, data output, and applications in target discovery and validation.
Core Mechanisms and Principles
RNA Interference (RNAi) Screening
RNAi utilizes small interfering RNAs (siRNAs) or short hairpin RNAs (shRNAs) delivered via transfection or viral transduction. These molecules guide the RNA-induced silencing complex (RISC) to complementary mRNA sequences, leading to degradation or translational repression. This results in knockdown of gene expression, which is typically incomplete and transient.
CRISPR-Cas9 Knockout Screening
CRISPR-Cas9 screens employ a single guide RNA (sgRNA) to direct the Cas9 endonuclease to a specific genomic DNA sequence. Cas9 creates a double-strand break, which is repaired by error-prone non-homologous end joining (NHEJ), often introducing insertions or deletions (indels) that disrupt the coding sequence of a gene, leading to a permanent knockout.
Quantitative Comparison of Key Parameters
Table 1: Head-to-Head Comparison of RNAi and CRISPR Screening Technologies
| Parameter | RNAi Screening (siRNA/shRNA) | CRISPR-Cas9 Knockout Screening |
|---|---|---|
| Molecular Target | mRNA | Genomic DNA |
| Effect on Gene | Knockdown (transcript degradation/translation block) | Knockout (frame-shift indels) |
| Efficacy (Typical Protein Reduction) | 70-90% (highly variable) | ~100% (in biallelic disrupted cells) |
| Duration of Effect | Transient (days to a week) | Permanent, heritable |
| Off-Target Effects | High (seed-sequence mediated; hundreds of potential targets) | Lower (20bp guide specificity; can be minimized with high-fidelity Cas9) |
| On-Target Efficacy Consistency | Low to Moderate (depends on reagent design/accessibility) | High (depends on sgRNA design and chromatin state) |
| Screening Library Size (Genome-wide) | ~3-5 shRNAs/siRNAs per gene | ~3-10 sgRNAs per gene |
| False Negative Rate | Higher (incomplete knockdown) | Lower (complete knockout) |
| False Positive Rate | Higher (off-targets, cytotoxicity) | Lower |
| Phenotype Penetrance | Variable, often muted | Typically strong |
| Suitability for Essential Gene Identification | Moderate (confounded by partial knockdown) | Excellent (clear, strong phenotypes) |
| Cost (Reagents & Sequencing) | Moderate | Moderate to High (depends on Cas9 delivery) |
Detailed Experimental Protocols
Protocol for a Pooled shRNA Knockdown Screen
Objective: Identify genes whose knockdown confers resistance to a chemotherapeutic agent. Workflow:
- Library Design & Cloning: Select a commercially available genome-wide lentiviral shRNA library (e.g., TRC, miR-E). Each shRNA is cloned in a lentiviral vector with a puromycin resistance marker.
- Virus Production: Produce lentivirus for the pooled shRNA library in HEK293T cells using standard packaging plasmids (psPAX2, pMD2.G).
- Cell Infection & Selection:
- Infect target cells (e.g., HeLa) at a low MOI (~0.3) to ensure most cells receive a single shRNA.
- Select transduced cells with puromycin (e.g., 2 µg/mL) for 48-72 hours.
- Challenge & Phenotypic Selection: Split cells into treatment (chemotherapeutic agent) and control (DMSO) arms. Culture for 14-21 population doublings to allow phenotype manifestation.
- Genomic DNA Extraction & PCR Amplification: Harvest cells. Isolate genomic DNA. Amplify the integrated shRNA barcode region using primers containing Illumina adaptor sequences.
- Next-Generation Sequencing (NGS): Pool PCR products and sequence on an Illumina platform.
- Bioinformatic Analysis: Map sequenced barcodes to the library manifest. Compare barcode read counts between treatment and control arms using specialized algorithms (e.g., RIGER, DESeq2) to identify significantly enriched or depleted shRNAs.
Protocol for a Pooled CRISPR-Cas9 Knockout Screen
Objective: Identify genes whose knockout confers sensitivity to a targeted inhibitor. Workflow:
- Cell Line Engineering: Stably express Cas9 in the target cell line via lentiviral transduction and blasticidin selection, or use a constitutive Cas9-expressing line.
- Library Design & Cloning: Use a genome-wide sgRNA library (e.g., Brunello, Brie). Each sgRNA is cloned into a lentiviral vector containing a puromycin resistance gene.
- Viral Production & Transduction: Produce lentiviral sgRNA library and transduce Cas9-expressing cells at an MOI of ~0.3. Select with puromycin.
- Phenotypic Selection: Split cells into treatment (inhibitor) and control arms. Passage cells for 14+ doublings.
- Genomic DNA Extraction & NGS Prep: Harvest pellets. Extract gDNA. Perform a two-step PCR: (i) amplify the sgRNA region, (ii) add Illumina indices and flow-cell adaptors.
- Sequencing & Analysis: Sequence pooled libraries. Align reads to the sgRNA library. Use model-based analysis (e.g., MAGeCK, BAGEL) to identify sgRNAs/genes significantly depleted (essential genes) or enriched (resistance genes) in the treatment arm.
Diagram 1: Comparative Workflows for Pooled RNAi and CRISPR Screens
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Functional Genomic Screens
| Item | Function & Description | Example Products/Providers |
|---|---|---|
| Genome-Wide Library | A pooled collection of shRNAs or sgRNAs targeting every gene in the genome. The foundation of the screen. | RNAi: Dharmacon TRC, Sigma MISSION, Cellecta. CRISPR: Broad Institute GPP (Brunello, Brie), Addgene, Synthego. |
| Lentiviral Packaging Plasmids | Required for producing replication-incompetent lentiviral particles to deliver sh/sgRNA libraries into target cells. | psPAX2 (packaging), pMD2.G (VSV-G envelope). |
| Cas9 Expression System | For CRISPR screens: provides the endonuclease. Can be delivered via stable cell line, plasmid, or mRNA. | lentiCas9-Blast (Addgene), all-in-one sgRNA/Cas9 lentiviral vectors, synthetic Cas9 protein. |
| Selection Antibiotics | To select for cells successfully transduced with the viral vector containing the resistance marker. | Puromycin, Blasticidin, Geneticin (G418). |
| NGS Library Prep Kit | For preparing the amplified sh/sgRNA barcodes for high-throughput sequencing. | Illumina TruSeq, NEBNext Ultra II. |
| Cell Line with High Viral Transduction Efficiency | Essential for achieving uniform library representation. Often requires specific growth properties. | HEK293T (for virus production), HeLa, K562, RPE1-hTERT. |
| Deep Sequencing Platform | To quantitatively count sh/sgRNA barcodes from pooled cell populations pre- and post-selection. | Illumina NextSeq, NovaSeq. |
| Bioinformatics Software | To statistically analyze sequencing counts and identify hit genes from screen data. | RNAi: RIGER, HiTSelect. CRISPR: MAGeCK, BAGEL, CRISPhieRmix. |
Diagram 2: Core Molecular Mechanisms of RNAi and CRISPR
Strategic Considerations and Emerging Applications
The choice between RNAi and CRISPR screens is context-dependent. RNAi remains useful for studying essential genes where complete knockout is lethal, allowing observation of hypomorphic phenotypes, and for in vivo screens where viral packaging size is limiting. CRISPR technology has largely supplanted RNAi for definitive loss-of-function studies, especially in identifying essential genes and drug targets with high confidence.
Furthermore, the CRISPR toolbox has expanded beyond knockout (CRISPRko) to include:
- CRISPR interference (CRISPRi): For reversible, transcript-specific knockdown without DNA cleavage.
- CRISPR activation (CRISPRa): For targeted gene overexpression screens.
- Base Editing & Prime Editing Screens: For precise nucleotide variant screening.
These modalities offer more nuanced comparisons to RNAi's knockdown phenotype.
In the context of advancing CRISPR-Cas9 knockout screen principles, the comparison with RNAi highlights a paradigm shift toward more precise, potent, and reliable genetic perturbation. CRISPR screens offer superior specificity, completeness, and consistency of gene inactivation, reducing false positives and negatives. However, RNAi retains niche applications. The selection of technology must align with the specific biological question, desired phenotype, and experimental constraints. The continued evolution of both platforms, particularly the expansion of CRISPR-based screening modalities, ensures functional genomics will remain a cornerstone of modern biological and therapeutic discovery.
This guide serves as a critical technical chapter within a broader thesis on CRISPR-Cas9 knockout screen principles. While the foundational mechanics of guide RNA libraries, Cas9 delivery, and sequencing analysis are well-established, the strategic selection of the screening paradigm is paramount to experimental success and biological insight. This document dissects the three cardinal factors—Phenotype, Gene Function, and Cell Type—that dictate the choice between arrayed and pooled screens, and the design of the screening assay itself.
Core Factors & Decision Framework
The interplay of the three factors determines the optimal screening strategy. Key quantitative considerations are summarized below.
Table 1: Decision Matrix for CRISPR Screen Selection
| Factor | Options / Considerations | Impact on Screen Design | Typical Throughput |
|---|---|---|---|
| Phenotype | Survival/Proliferation | Pooled, positive/negative selection | High (Genome-wide) |
| Fluorescence (FACS) | Pooled or Arrayed | Medium to High | |
| Imaging (Morphology, Spatial) | Arrayed | Low to Medium | |
| Transcriptional (scRNA-seq) | Pooled (Perturb-seq, CROP-seq) | Medium | |
| Gene Function | Genome-wide Discovery | Pooled | High (50k+ guides) |
| Focused Library (Pathway, Druggable) | Pooled or Arrayed | Medium (5k-20k guides) | |
| Custom Hypothesis Testing | Arrayed | Low (<5k guides) | |
| Cell Type | Adherent, Robustly Proliferating | Compatible with all screens | N/A |
| Non-Adherent/Suspension | Favors pooled screens | N/A | |
| Primary/Non-dividing | Requires specialized delivery (e.g., nucleofection); often arrayed | Low | |
| Differentiated/Stem | May require inducible Cas9; phenotype-dependent | Variable |
Detailed Experimental Protocols
Protocol 1: Pooled CRISPR Knockout Screen for Essential Genes (Survival Phenotype)
- Objective: Identify genes essential for cell proliferation/survival in a given cell line.
- Materials: See "Scientist's Toolkit" (Table 2).
- Method:
- Library Transduction: Transduce the target cell line (e.g., A549) at a low MOI (~0.3) with the lentiviral pooled sgRNA library (e.g., Brunello) to ensure >95% of cells receive a single guide. Include a coverage of at least 500 cells per sgRNA.
- Selection & Expansion: Treat cells with puromycin (1-2 µg/mL) for 7 days to select for transduced cells. Allow cells to proliferate for an additional 14-21 population doublings.
- Sample Harvesting: Harvest genomic DNA (gDNA) at the initial timepoint (T0) post-selection and at the final endpoint (Tfinal) using a mass-culture method.
- sgRNA Amplification & Sequencing: Amplify the integrated sgRNA cassette from gDNA via a two-step PCR. The first PCR (25 cycles) amplifies the region from bulk gDNA; the second PCR (10-12 cycles) adds Illumina adaptors and sample barcodes.
- Analysis: Sequence PCR products on an Illumina HiSeq. Align reads to the library reference. Calculate fold-depletion of each sgRNA from T0 to Tfinal using a robust statistical model (e.g., MAGeCK or BAGEL2) to identify significantly depleted essential genes.
Protocol 2: Arrayed CRISPR Knockout Screen for High-Content Imaging Phenotype
- Objective: Quantify changes in subcellular morphology (e.g., nuclear fragmentation, cytoskeletal rearrangement) upon gene knockout.
- Materials: See "Scientist's Toolkit" (Table 2).
- Method:
- Reverse Transfection: Seed cells (e.g., U2OS) in 384-well imaging plates. Using a liquid handler, co-transfect pre-arrayed synthetic sgRNAs (50nM) and Cas9 ribonucleoprotein (RNP) complexes using a lipid-based transfection reagent.
- Incubation: Incubate cells for 72-96 hours to allow for protein turnover and phenotype manifestation.
- Fixation and Staining: Fix cells with 4% PFA, permeabilize with 0.1% Triton X-100, and stain for relevant markers (e.g., DAPI for nuclei, Phalloidin for actin).
- Image Acquisition & Analysis: Acquire images on a high-content confocal imager (e.g., Opera Phenix). Use integrated software (e.g., Harmony) to segment cells and extract ~500 morphological features per cell. Perform Z-score normalization per plate and use a Mann-Whitney U test to compare each knockout well to negative control wells.
Visualizing the Screening Workflow & Pathway Integration
Diagram Title: Decision Flow for CRISPR Screen Selection
Diagram Title: Integrating Pathway Knowledge into Focused Screen Design
The Scientist's Toolkit
Table 2: Essential Research Reagents & Solutions
| Item | Function & Application | Example/Supplier |
|---|---|---|
| Genome-Wide sgRNA Library | Pre-designed, cloned lentiviral pools targeting all human genes. Enables discovery screens. | Brunello, TorontoKO (Addgene) |
| Focused sgRNA Library | Subset library targeting specific gene families (kinases, GPCRs) or pathways. Lowers cost & complexity. | Dharmacon CRISPRko sub-libraries |
| Arrayed sgRNA Collection | Individual sgRNAs in multi-well plates. Enables reverse transfection & complex assays. | Horizon Discovery Arrayed文库 |
| Lentiviral Packaging Mix | Plasmids (psPAX2, pMD2.G) for producing infectious, replication-incompetent lentivirus. | Standard third-generation system |
| Cas9 Expression System | Stable cell line (Cas9-expressing) or delivery format (plasmid, mRNA, RNP). | ToolGen Cas9 cell line; IDT Alt-R S.p. Cas9 Nuclease V3 |
| Transfection Reagent (Lipid) | For arrayed screens; delivers synthetic sgRNAs and Cas9 RNP into cells. | Lipofectamine CRISPRMAX (Invitrogen) |
| Nucleofection Kit | Electroporation-based delivery for hard-to-transfect cells (primary, suspension). | Lonza 4D-Nucleofector Kits |
| Next-Gen Sequencing Kit | For pooled screen deconvolution; prepares sgRNA amplicons for Illumina sequencing. | Illumina Nextera XT DNA Library Prep Kit |
| High-Content Imaging System | Automated microscope + software for phenotypic analysis in arrayed screens. | PerkinElmer Opera Phenix, Molecular Devices ImageXpress |
| Analysis Software | Statistical packages for identifying enriched/depleted genes from NGS data. | MAGeCK, BAGEL2 (open source) |
Within CRISPR-Cas9 functional genomics research, a core thesis posits that systematic knockout screens reveal genetic dependencies—genes essential for cellular fitness. Integrating these dependency profiles with transcriptomic and proteomic data is critical for understanding the mechanistic basis of vulnerability, distinguishing driver from passenger effects, and identifying druggable pathways. This whitepaper provides a technical guide for this multi-omics integration, framing methodologies within the context of advancing CRISPR screen principle research for target discovery in oncology and beyond.
Core Data Types and Quantitative Landscape
The integration correlates three primary data modalities, each with characteristic scales and outputs from modern platforms.
Table 1: Core Multi-Omics Data Modalities for Integration with Genetic Dependencies
| Data Type | Primary Technology | Typical Scale (Per Sample) | Key Output Metric | Relevance to Dependency |
|---|---|---|---|---|
| Genetic Dependency | CRISPR-Cas9 Pooled Screen | 500-20,000 genes | CERES score, DepMap Chronos score (≈ -2 to +2) | Direct measure of gene essentiality. Negative score indicates loss of fitness upon knockout. |
| Transcriptomic | Bulk or Single-Cell RNA-Seq | 20,000 genes | TPM, FPKM, Log2(Counts) | Steady-state mRNA levels. Can reveal overexpression in dependent cell lines or compensatory pathways. |
| Proteomic | Mass Spectrometry (LF, TMT) or RPPA | 3,000 - 10,000 proteins | Log2(Intensity), iBAQ | Functional effector levels. Post-translational modifications (e.g., phosphorylation) indicate pathway activity. |
Foundational Experimental Protocols
Generating Genetic Dependency Data via CRISPR-Cas9 Screens
Protocol: Genome-wide Pooled Knockout Screen (adapted from DepMap/Score methodology)
- Library Design: Use the Brunello or similar genome-wide sgRNA library (≈4-6 sgRNAs/gene, 80,000 total sgRNAs).
- Viral Transduction: Transduce a Cas9-expressing cell line (e.g., derived from a cancer model) at low MOI (<0.3) to ensure single integration. Select with puromycin for 3-5 days.
- Passaging & Harvest: Passage cells for a minimum of 14 population doublings. Harvest genomic DNA at the initial (T0) and final (Tend) time points.
- Amplification & Sequencing: PCR-amplify integrated sgRNA sequences with barcoded primers. Perform deep sequencing (Illumina).
- Analysis: Align reads to the library reference. Calculate gene-level essentiality scores (e.g., CERES) using tools like MAGeCK or BAGEL2, which account for sgRNA efficiency and copy-number effects.
Generating Correlative Omics Profiles
Protocol: Bulk RNA-Sequencing for Transcriptomics
- Sample Prep: Harvest cell pellets from the same cell line used in dependency screens, ideally under matched culture conditions.
- Library Prep: Isolate total RNA (RIN > 8.5). Use poly-A selection for mRNA. Prepare libraries with strand-specific kits (e.g., Illumina TruSeq).
- Sequencing: Sequence on an Illumina platform to a depth of 30-50 million paired-end reads per sample.
- Analysis: Align to a reference genome (STAR, HISAT2). Quantify gene counts (featureCounts). Normalize (TPM, DESeq2) and transform (log2(TPM+1)).
Protocol: Data-Independent Acquisition (DIA) Mass Spectrometry for Proteomics
- Sample Lysis & Digestion: Lyse cells in RIPA buffer. Reduce, alkylate, and digest proteins with trypsin.
- Peptide Clean-up: Desalt using C18 solid-phase extraction.
- LC-MS/MS Analysis: Separate peptides on a nano-flow LC system coupled to a high-resolution tandem mass spectrometer (e.g., timsTOF, Orbitrap) operating in DIA mode.
- Analysis: Process raw files using spectral library-based tools (Spectronaut, DIA-NN) or library-free approaches. Report protein abundances as log2 intensities.
Data Integration Methodologies
Correlation Analysis
The fundamental approach calculates pairwise correlations (Spearman's ρ) between dependency scores of a gene of interest and the expression levels of all other genes/proteins across a panel of cell lines (e.g., Cancer Cell Line Encyclopedia - CCLE).
Workflow: From Raw Data to Integrated Insights
Diagram 1: Core Multi-Omics Integration Workflow
Pathway and Network Analysis
Correlation results are interpreted through pathway over-representation analysis (ORA) or gene set enrichment analysis (GSEA) using databases like MSigDB, Reactome, or KEGG. Protein-protein interaction networks (from STRING) can be overlaid with correlation z-scores.
Logical Flow for Mechanistic Hypothesis Generation
Diagram 2: From Correlation to Mechanistic Hypothesis
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Resources for Multi-Omics Integration Studies
| Item | Supplier/Resource | Function in Workflow |
|---|---|---|
| Genome-wide sgRNA Library (Brunello) | Addgene (Kit #73179) | Provides pre-validated sgRNA sequences for targeting human genes in CRISPR screens. |
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Addgene (#12260, #12259) | Essential for producing lentiviral particles to deliver sgRNA libraries. |
| Puromycin Dihydrochloride | Thermo Fisher (A1113803) | Selective antibiotic for cells post-transduction with sgRNA vectors. |
| TruSeq Stranded mRNA Library Prep Kit | Illumina (20020594) | Standardized kit for preparing sequencing libraries from poly-A RNA. |
| Trypsin, Sequencing Grade | Promega (V5111) | Protease for digesting proteins into peptides for mass spectrometry analysis. |
| TMTpro 16plex Label Reagent Set | Thermo Fisher (A44520) | Isobaric tags for multiplexed quantitative proteomics across many samples. |
| DepMap Public Data Portal (23Q4) | Broad Institute | Primary source for pre-computed dependency scores (Chronos) and omics data for 1000+ cell lines. |
| CCLE Data Portal | Broad Institute | Source for harmonized transcriptomic (RNA-seq) and proteomic (RPPA) data for cancer cell lines. |
Conclusion
CRISPR-Cas9 knockout screens have revolutionized functional genomics by enabling systematic, genome-wide interrogation of gene function. This guide has walked through the core principles, methodological execution, critical optimization steps, and comparative landscape of this powerful technology. For biomedical research and drug discovery, CRISPR screens offer an unparalleled path to identifying genetic dependencies, novel therapeutic targets, and mechanisms of drug action and resistance. Future directions point toward more sophisticated in vivo and organoid models, higher-fidelity editing systems to reduce artifacts, and the integration of single-cell readouts to dissect complex cellular phenotypes. As the technology matures, its role in translating genetic insight into clinical innovation will only continue to expand.