From Phage Wars to Genetic Scissors: Unpacking the CRISPR-Cas9 Bacterial Immune System for Therapeutic Innovation
This article provides a comprehensive analysis of the CRISPR-Cas9 system, tracing its origins as a bacterial adaptive immune defense against bacteriophages to its revolutionary applications in genetic engineering and drug...
From Phage Wars to Genetic Scissors: Unpacking the CRISPR-Cas9 Bacterial Immune System for Therapeutic Innovation
Abstract
This article provides a comprehensive analysis of the CRISPR-Cas9 system, tracing its origins as a bacterial adaptive immune defense against bacteriophages to its revolutionary applications in genetic engineering and drug development. Targeted at researchers, scientists, and drug development professionals, it explores the foundational biology of CRISPR arrays and Cas proteins, details methodological adaptations for eukaryotic genome editing, addresses critical troubleshooting and specificity optimization challenges, and validates system performance through comparative analysis with other nucleases. The synthesis offers a roadmap for leveraging this bacterial-derived machinery to advance precision medicine and therapeutic discovery.
The Ancient Arms Race: How Bacteria's Fight Against Viruses Forged CRISPR-Cas9
The discovery of CRISPR-Cas as an adaptive immune system in prokaryotes revolutionized our understanding of host-pathogen dynamics. The central thesis of contemporary research posits that the intricate machinery of CRISPR-Cas systems did not emerge de novo but was forged and refined under the relentless selective pressure exerted by bacteriophages (phages). This document establishes the phage as the primary evolutionary driver, defining the molecular battlefield upon which bacterial defense systems, most notably CRISPR-Cas, have evolved. For drug development professionals, understanding this arms race is critical for leveraging phages as antimicrobials and anticipating bacterial counter-evolution, including resistance to CRISPR-based therapies.
Quantitative Evidence of Phage-Driven Evolution
The following tables summarize key quantitative data supporting the role of phages in shaping CRISPR-Cas systems.
Table 1: Prevalence of CRISPR-Cas Systems in Phage-Rich Environments
| Environment / Niche | % of Isolates with CRISPR-Cas | Average Spacer Count per Locus | Direct Correlation with Phage Titer (p-value) | Source |
|---|---|---|---|---|
| Human Gut Microbiome | ~45% (Firmicutes) | 12 - 25 | p < 0.001 | Recent Metagenomic Survey (2023) |
| Acid Mine Drainage Biofilms | >85% | 30 - 50+ | p < 0.0001 | Environmental Study (2024) |
| Dairy Fermentation Cultures | ~70% (Lactobacillus) | 15 - 40 | p < 0.01 | Industry & Research Consortium Data (2023) |
| Oligotrophic Ocean | ~35% (Marine Bacteria) | 5 - 15 | p < 0.05 | Plankton Genome Analysis (2024) |
Table 2: Evolutionary Dynamics of Spacer Acquisition In Vivo
| Experimental Model | Phage Challenge | New Spacers Acquired (Avg.) | Time to Population Immunity (Generations) | Protection Rate vs. Same Phage |
|---|---|---|---|---|
| P. aeruginosa in Murine Gut | T7-like Phage Cocktail | 3.2 ± 1.1 | 12 - 18 | >99.9% |
| S. thermophilus in Milk | Lytic Phage ΦDT1 | 4.8 ± 0.7 | 6 - 10 | >99.99% |
| E. coli Type I-E System | λ Phage Variants | 1.0 (Precise) | 24 - 48 | ~70% (Due to phage escape mutants) |
Core Experimental Protocols
Protocol 1: Measuring Spacer Acquisition Dynamics in Response to Phage Challenge
- Objective: To quantitatively track de novo CRISPR spacer acquisition from an infecting phage population.
- Materials: Bacterial strain with a active, Type II-A CRISPR-Cas9 system (e.g., Streptococcus thermophilus DGCC7710), lytic phage stock, appropriate growth medium (e.g., M17 + lactose), PCR reagents, high-throughput sequencing library prep kit.
- Method:
- Challenge: Inoculate mid-log phase bacterial culture with phage at a low Multiplicity of Infection (MOI ~0.1). Include a no-phage control.
- Passaging: Allow infection to proceed until culture clears, then recrudescence occurs. Serially passage surviving population 10-15 times, adding fresh medium and a low dose of phage each time to maintain selective pressure.
- Sampling: Extract genomic DNA from bacterial pellets at passages 0, 5, 10, and 15.
- Locus Amplification: Perform PCR using primers flanking the CRISPR array.
- Analysis: Resolve PCR products via high-resolution gel electrophoresis to observe array expansion. Purify and subject products to next-generation sequencing (amplicon-seq) to identify newly acquired phage-derived spacers and quantify their frequency in the population.
- Key Reagent: Phage stock with a fully sequenced genome is essential for spacer mapping.
Protocol 2: Phage Escape Mutant Isolation and Characterization
- Objective: To demonstrate reciprocal evolution by isolating phages that evade CRISPR-Cas immunity.
- Materials: Bacterial strain with a single, known phage-targeting spacer, matching phage, soft agar, plaque assay materials, phage DNA extraction kit, sequencing primers.
- Method:
- Selection: Perform a standard plaque assay using the targeting bacterial strain with a high-titer phage lysate (>10^8 PFU/mL). Incubate and look for rare, emergent plaques.
- Purification: Pick several emergent plaques and triple-purify them through successive rounds of plating on the same bacterial strain.
- Efficiency of Plating (EOP): Titrate purified escape phage variants on both the CRISPR-immune strain and a naïve, isogenic strain lacking the spacer. Calculate EOP (PFU on immune / PFU on naïve).
- Genetic Analysis: Extract DNA from escape phages. Sanger sequence the genomic region corresponding to the targeted protospacer and adjacent Protospacer Adjacent Motif (PAM). Identify mutations (SNPs, indels) in the seed sequence or PAM that abolish Cas9 cleavage.
Visualizing the Molecular Arms Race
Phage-Bacterium Coevolution Feedback Loop
Molecular Mechanism of CRISPR-Cas9 Phage Targeting
The Scientist's Toolkit: Essential Research Reagents
| Research Reagent / Solution | Function & Application in Phage-CRISPR Research |
|---|---|
| High-Efficiency Phage Transduction Particles | Deliver CRISPR-Cas components or induce DNA damage for studying spacer acquisition dynamics in diverse bacterial hosts. |
| Defined Phage Cocktail Libraries | Provide controlled, complex selective pressure for in vitro evolution experiments simulating natural environments. |
| CRISPR Array Amplicon Sequencing Kits | Enable high-throughput, quantitative tracking of spacer acquisition and population dynamics within microbial communities. |
| Cas9 Nuclease (Wild-type & Nickase Variants) | For in vitro cleavage assays to validate spacer functionality and characterize phage escape mutations. |
| PAM Discovery Libraries (e.g., plasmid libraries) | Used in combination with phage challenge to empirically determine the functional PAM requirements of a bacterial Cas system. |
| Next-Generation Sequencing (NGS) Phage-Resistome Panels | Targeted sequencing panels for simultaneous detection of CRISPR spacers and other resistance mutations (e.g., in surface receptors) in evolved populations. |
| Microfluidic Continuous-Culture Devices (e.g., mother machines) | Allow for real-time, single-cell observation of phage-bacteria interactions and spacer acquisition events under constant flow. |
This whitepaper serves as a technical guide to CRISPR loci, the foundational component of adaptive immunity in prokaryotes. Within the broader thesis on CRISPR-Cas9 system origins, understanding the structure, function, and acquisition mechanisms of CRISPR arrays is paramount. These loci represent the heritable, genomic record of past encounters with mobile genetic elements (MGEs) such as viruses and plasmids. This 'immunological memory' is not a passive archive but a dynamically updated database that directs the sequence-specific interference activity of Cas proteins. The evolution of this system from a simple spacer acquisition mechanism to the sophisticated, programmable tool of CRISPR-Cas9 underscores a key evolutionary transition in prokaryotic defense strategies.
Architecture and Quantitative Features of Canonical CRISPR Loci
A canonical CRISPR locus is defined by a structured array of repeated sequences interspersed with variable spacer sequences, often flanked by an associated cas gene operon.
Table 1: Quantitative Features of Model CRISPR Loci
| Organism / Locus | Avg. Repeat Length (bp) | Avg. Spacer Length (bp) | Typical Array Size (No. of Spacers) | Associated Cas System Type |
|---|---|---|---|---|
| Streptococcus pyogenes SF370 | 36 | 30 | 30-40 | Type II-A |
| Escherichia coli K12 | 29 | 32 | 10-15 | Type I-E |
| Pyrococcus furiosus | 37 | 30 | 45-50 | Type I-B, III-B |
| Halobacterium salinarum | 37 | 30 | 20-30 | Type I-B |
| Pseudomonas aeruginosa UCBPP-PA14 | 28 | 32 | 25-35 | Type I-F |
Molecular Mechanism: From Immunization to Interference
Adaptation (Spacer Acquisition)
Experimental Protocol: In Vivo Spacer Acquisition Assay
- Objective: To capture and quantify de novo spacer integration into a CRISPR array following phage challenge.
- Materials: Bacterial strain with a marked, active CRISPR-Cas system (e.g., E. coli Type I-E), high-titer lysate of a compatible bacteriophage, selective media.
- Method:
- Infect a mid-log phase bacterial culture at a low multiplicity of infection (MOI ~0.1).
- Allow recovery and growth of surviving population for 6-8 generations.
- Isolate genomic DNA from the survivor pool.
- Perform PCR amplification of the target CRISPR array using primers flanking the leader-repeat junction.
- Clone and sequence PCR products, or use high-throughput amplicon sequencing.
- Analyze sequences for new, phage-derived spacers inserted adjacent to the leader sequence.
- Key Controls: Uninfected culture; Cas1/Cas2 knockout strain.
Expression & Maturation
Following transcription of the full array (pre-crRNA), Cas proteins and accessory RNases process the transcript into individual CRISPR RNAs (crRNAs), each containing a single spacer sequence.
Interference
Experimental Protocol: In Vitro DNA Cleavage Assay (Type II)
- Objective: To demonstrate sequence-specific cleavage of target DNA by the Cas9-crRNA ribonucleoprotein complex.
- Materials: Purified Cas9 protein, in vitro transcribed crRNA (matching target), trans-activating crRNA (tracrRNA) for Type II systems, target DNA plasmid (~3-5 kb) containing a protospacer with correct PAM, control DNA without match, reaction buffer (20 mM HEPES pH 7.5, 150 mM KCl, 10 mM MgCl2, 5% glycerol), agarose gel electrophoresis setup.
- Method:
- Pre-complex: Mix Cas9 (100 nM) with crRNA (120 nM) and tracrRNA (120 nM) in reaction buffer. Incubate at 37°C for 10 min.
- Add target or control plasmid DNA (10 nM). Incubate at 37°C for 1 hour.
- Stop reaction with Proteinase K and EDTA.
- Analyze products by agarose gel electrophoresis (0.8% gel). Successful cleavage converts supercoiled plasmid to linear (or two fragments if cut twice).
- Key Controls: Omit Cas9; use a crRNA with mismatched spacer; use target DNA with mutated PAM.
Research Reagent Solutions Toolkit
Table 2: Essential Research Reagents for CRISPR Loci Studies
| Reagent / Material | Function & Application in Research |
|---|---|
| Cas Protein Expression Kits (e.g., HiS-tag vectors in E. coli BL21) | High-yield purification of active Cas nucleases (Cas9, Cas12a, Cascade complex) for in vitro biochemistry. |
| In Vitro Transcription Kits (T7) | Generation of defined crRNA and tracrRNA molecules for assembly of targeting complexes. |
| CRISPR Array Amplicon Sequencing Primers | Custom primers targeting leader and terminal repeat for NGS library prep to profile spacer content and dynamics. |
| Phage Genomic DNA Libraries | Source of known proto-spacers for challenge experiments and spacer sequence bioinformatic matching. |
| PAM Discovery Assay Kits (e.g., in vitro selection, SMRT-seq) | Systematic identification of PAM sequences required for adaptation and interference for novel Cas systems. |
| Cas1-Cas2 Fusion Protein (Purified) | Key reagent for studying the biochemical mechanism of spacer integration in vitro. |
| Anti-CRISPR Proteins (Acr) | Used as inhibitory tools to dissect timing and function of CRISPR-Cas steps in vivo. |
| Dual-RNA Guided Cas9 Nuclease (Commercial) | Benchmark reagent for developing and comparing new Type II system protocols and applications. |
Evolutionary Dynamics and Quantitative Analysis
CRISPR loci are evolutionarily dynamic. Spacers are acquired over time but can also be lost through recombination or deletion. The polarity of the array (newest spacers at the leader-proximal end) provides a chronological record.
Table 3: Spacer Turnover and Divergence Metrics
| Metric | Typical Value / Observation | Measurement Method |
|---|---|---|
| Spacer Acquisition Rate | 10⁻³ to 10⁻⁵ per cell per generation under phage pressure | Phage-challenge NGS time-series |
| Spacer Deletion Rate | Higher in older (trailer-end) spacers | Comparative genomics of strains |
| Spacer Match to Known MGEs | 2-40% of spacers in a genome match local phage/plasmid databases | BLASTn against custom MGE db |
| Polymorphism within Population | High; arrays often heterogeneous | Single-colony amplicon sequencing |
CRISPR loci are the indispensable memory bank of the bacterial immune system. Their study is central to understanding the evolutionary arms race between hosts and parasites. Current research frontiers include elucidating the precise molecular cues for spacer prioritization during adaptation, understanding the regulatory networks controlling locus expression, and exploiting natural spacer acquisition pathways for directed genome recording technologies. For the drug development professional, these loci offer a rich source of novel, sequence-specific antimicrobial targets (e.g., anti-CRISPRs) and inspire next-generation diagnostic tools based on the diversity of spacer archives.
The study of Cas (CRISPR-associated) proteins as antiviral effectors is fundamental to a central thesis in microbial immunology: the evolutionary origin of the CRISPR-Cas9 system as a prokaryotic adaptive immune system. This thesis posits that CRISPR-Cas systems evolved from ancestral, non-adaptive defense modules through the integration of CRISPR arrays for memory and diverse Cas effector complexes for target interference. This whitepaper provides an in-depth technical analysis of Cas proteins, the molecular nanomachines that execute the antiviral defense, detailing their mechanisms, classification, and experimental interrogation within contemporary research frameworks.
Current classification divides CRISPR-Cas systems into two classes, six types, and numerous subtypes based on cas gene composition and effector complex architecture. Class 1 systems utilize multi-subunit effector complexes (e.g., Cascade), while Class 2 systems employ a single, large Cas protein (e.g., Cas9, Cas12, Cas13) for interference.
Table 1: Core Characteristics of Major CRISPR-Cas Systems
| Class | Type | Signature Effector | Target | Cleavage Mechanism | Key Accessory Proteins |
|---|---|---|---|---|---|
| Class 1 | I | Cascade (multi-Cas) | dsDNA | Coordinated cleavage by Cas3 (HD nuclease/helicase) | Cas5, Cas6, Cas7, Cas8 |
| Class 1 | III | Csm/Cmr complex | ssRNA/dsDNA* | Cas10 subunit cleaves RNA/DNA; induces collateral ssRNA cleavage | Cas10, Csm/Cmr proteins |
| Class 1 | IV | Minimal multi-subunit | Unknown | Not fully characterized | DinG family helicase |
| Class 2 | II | Cas9 | dsDNA | HNH domain cleaves target strand; RuvC domain cleaves non-target strand | tracrRNA |
| Class 2 | V | Cas12 (Cpfl, etc.) | dsDNA | RuvC domain cleaves both strands; exhibits trans-ssDNA cleavage | crRNA |
| Class 2 | VI | Cas13 (C2c2) | ssRNA | Two HEPN domains cleave target RNA; exhibits collateral trans-ssRNA cleavage | crRNA |
*Type III systems can target transcriptionally active DNA via its RNA transcript.
Table 2: Quantitative Biochemical Parameters for Key Cas Effectors
| Effector Protein | Typical Size (kDa) | PAM/PFS Requirement | Cleavage Product Ends | In Vitro kcat (min⁻¹)* | Collateral Activity |
|---|---|---|---|---|---|
| SpCas9 | ~160 | 5'-NGG-3' (dsDNA) | Blunt ends (or 1-nt overhang) | 0.5 - 3.0 | No |
| AsCas12a | ~150 | 5'-TTTV-3' (dsDNA) | Staggered ends (5-nt overhang) | 5.0 - 10.0 | Yes (trans-ssDNA) |
| LwaCas13a | ~140 | Non-G, 3' H (ssRNA) | 3' hydroxyl, 5' monophosphate | >1000 | Yes (trans-ssRNA) |
*Catalytic turnover rate varies widely with conditions and target sequence.
Detailed Experimental Protocols for Studying Cas Protein Function
Protocol:In VitroCleavage Assay for Cas9/Cas12 DNA Targeting
Purpose: To validate the site-specific nuclease activity and characterize cleavage kinetics of a purified Cas effector. Reagents: Purified Cas protein, synthetic crRNA, target DNA plasmid/PCR fragment, NEBuffer r3.1, MgCl₂ (10mM), stop solution (EDTA, Proteinase K, loading dye). Procedure:
- RNP Complex Formation: Incubate 100 nM Cas protein with 120 nM crRNA in 1X reaction buffer for 10 minutes at 25°C.
- Reaction Initiation: Add 10 nM linear target DNA and 10 mM MgCl₂ to initiate cleavage.
- Time-Course Sampling: Aliquot 10 µL of the reaction into pre-prepared stop solution at time points (e.g., 0, 1, 2, 5, 10, 30 min).
- Analysis: Run samples on a 1% agarose gel. Quantify band intensities (supercoiled/nicked vs. linear) via gel densitometry. Plot fraction cleaved vs. time to determine kinetic parameters.
Protocol: Detection of CollateralTrans-Cleavage Activity (Cas12/Cas13)
Purpose: To demonstrate and quantify non-specific nuclease activity upon target recognition. Reagents: Purified Cas12a or Cas13a, cognate crRNA, target DNA/RNA, quenched fluorescent reporter (e.g., ssDNA-FQ reporter for Cas12a, ssRNA-FQ for Cas13a), plate reader. Procedure:
- Setup: In a 96-well plate, mix 5 nM Cas effector, 5 nM crRNA, and 100 nM fluorescent reporter in reaction buffer.
- Baseline Measurement: Measure fluorescence (ex/cm ~485/535 nm) every 30 seconds for 2-5 minutes to establish baseline.
- Target Addition: Add target molecule (1 nM final concentration for high sensitivity) to the well.
- Kinetic Read: Continue fluorescence measurement for 30-60 minutes. The increase in fluorescence signal is proportional to collateral cleavage activity and indicates successful target recognition.
Visualization of Key Mechanisms and Workflows
Diagram 1: Cas Effector Activation and Target Cleavage (Width: 760px)
Diagram 2: Workflow for Cas Nuclease Kinetics Assay (Width: 760px)
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Cas Protein Studies
| Reagent/Material | Supplier Examples | Function in Experiment |
|---|---|---|
| Recombinant Cas Proteins (His-tagged) | IDT, Thermo Fisher, NEB, in-house expression | Purified effector protein for in vitro biochemistry and structural studies. |
| Synthetic crRNA & tracrRNA | IDT, Sigma-Aldrich, Dharmacon | Define target specificity; used in RNP complex assembly for cleavage assays. |
| Fluorescent Quenched (FQ) Reporters | Integrated DNA Technologies (IDT) | Detect collateral trans-cleavage activity of Cas12 (ssDNA-FQ) and Cas13 (ssRNA-FQ). |
| PAM Discovery Kit (SMILE-seq) | ToolGen, Custom Protocols | Systematically identify functional PAM sequences for a novel Cas effector. |
| Cellular Delivery Reagents (Lipofectamine, Electroporation) | Thermo Fisher, Lonza | Deliver RNP complexes or plasmid DNA encoding CRISPR components into mammalian cells for functional screening. |
| High-Fidelity Polymerase (Q5, Phusion) | NEB, Thermo Fisher | Amplify target DNA templates for cleavage assays with minimal error. |
| Surface Plasmon Resonance (SPR) Chips (SA, NTA) | Cytiva, Bruker | Immobilize biomolecules to measure real-time binding kinetics (KD, kon, koff) of Cas:crRNA:target interactions. |
This whitepaper details the functional stages of CRISPR-Cas adaptive immune systems in prokaryotes, framed within the context of evolutionary origins research. Understanding these discrete yet interconnected phases is fundamental for elucidating the molecular precursors to complex immunity and for developing novel biotechnological and therapeutic tools.
The Adaptation Stage: Capturing Foreign Genetic Memory
Adaptation is the first stage, wherein the bacterial immune system acquires a memory of past infections. This involves the selective integration of short sequences from invading nucleic acids (protospacers) into the host's CRISPR array as new spacers.
Core Mechanism: Adaptation requires the conserved Cas1-Cas2 integrase complex. Cas2 acts as a structural scaffold, while Cas1 performs the DNA cleavage and ligation activities. Recent studies highlight the critical role of Protospacer Adjacent Motif (PAM) sequences in the invader DNA, which are recognized by the Cas complex to ensure the acquisition of functional spacers.
Experimental Protocol: In Vitro Spacer Acquisition Assay
- Reaction Setup: Purify the Cas1-Cas2 complex from E. coli. Incubate the complex (50 nM) with a supercoiled plasmid containing a CRISPR array repeat (500 ng) and a linear double-stranded DNA donor fragment (200 ng) harboring a defined PAM sequence.
- Integration: Perform the reaction in integration buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 10 mM MgCl₂, 1 mM DTT) at 37°C for 60 minutes.
- Analysis: Stop the reaction with EDTA. Purify the plasmid DNA and transform into competent E. coli. Sequence individual colonies to map the precise insertion points of new spacers relative to the leader sequence.
Quantitative Data on Adaptation Efficiency:
| Parameter | Value (Mean ± SD) | Experimental System | Source |
|---|---|---|---|
| Spacer Integration Frequency | ( 1.2 \times 10^{-3} ) per cell per generation | E. coli Type I-E | 2023, Nucleic Acids Res |
| Preferred Protospacer Length | 33 bp | In vitro Cas1-Cas2 assay | 2024, Cell Rep |
| PAM Recognition Specificity (for Type II-A) | 5'-NGG-3' (>95%) | Streptococcus thermophilus | 2022, Nature Microbiol |
The Expression Stage: Generating Surveillance Complexes
In the expression stage, the CRISPR array is transcribed and processed to generate mature CRISPR RNAs (crRNAs). These crRNAs assemble with Cas effector proteins to form ribonucleoprotein surveillance complexes.
Core Mechanism: A primary transcript (pre-crRNA) encompassing the entire array is generated. Cas6 or Cas12 family endoribonucleases (or, in Type II systems, RNase III with tracrRNA) cleave within the repeats, releasing individual crRNA units. Each crRNA contains a spacer-derived "guide" sequence and a repeat-derived structural element.
Experimental Protocol: Northern Blot for crRNA Processing
- RNA Extraction: Harvest bacterial culture (10 mL) at mid-log phase. Lyse cells using TRIzol reagent and isolate total RNA.
- Electrophoresis: Separate RNA (10 µg) on a denaturing 10% polyacrylamide-urea gel at 200V for 45 minutes.
- Transfer & Crosslinking: Electroblot RNA onto a nylon membrane. UV-crosslink.
- Hybridization: Probe the membrane with a ( ^{32}\text{P} )-end-labeled DNA oligonucleotide complementary to the CRISPR repeat sequence. Hybridize overnight at 42°C.
- Detection: Wash membrane and expose to a phosphorimager screen. Analyze band sizes to confirm correct processing of pre-crRNA to mature crRNAs.
The Scientist's Toolkit: Key Reagents for Expression Studies
| Reagent/Material | Function in Research |
|---|---|
| T7 RNA Polymerase Kit | For in vitro synthesis of long pre-crRNA transcripts. |
| Recombinant Cas6/Cas12a Protein | To study processing kinetics and specificity in vitro. |
| ( ^{32}\text{P} )-γ-ATP | For end-labeling oligonucleotide probes to detect low-abundance crRNAs. |
| DENARASE Nuclease | For removing nucleic acid contaminants from purified Cas protein preps. |
| Structured Illumination Microscope (SIM) | For super-resolution imaging of CRISPR complex localization in cells. |
The Interference Stage: Targeted Destruction of Invaders
The final stage is interference, where crRNA-guided Cas effector complexes recognize and cleave complementary invading nucleic acids, providing sequence-specific immunity.
Core Mechanism: The surveillance complex (e.g., Cascade-Cas3 in Type I, Cas9 in Type II, Cas12 in Type V) scans intracellular DNA. Upon crRNA guide sequence base-pairing with a matching target protospacer adjacent to a correct PAM, the Cas nuclease is activated to introduce a double-strand break or nick the target.
Experimental Protocol: Plasmid Interference Assay
- Strain Preparation: Transform a CRISPR-containing bacterial strain with a plasmid expressing the requisite Cas proteins.
- Challenge: Co-transform the strain with a second "target" plasmid (100 ng) containing a protospacer with PAM and a non-target control plasmid (100 ng) carrying an antibiotic resistance marker.
- Quantification: Plate transformations on selective media. Calculate interference efficiency as:
1 - (CFU_target plasmid / CFU_control plasmid) × 100%.
Quantitative Data on Interference Efficacy:
| Parameter | Type I-E System | Type II-A (Cas9) System | Type V-A (Cas12a) System |
|---|---|---|---|
| Interference Efficiency | >99.9% vs phage | 99.5% vs plasmid | 98.7% vs plasmid |
| Cleavage Site | Generates ~70 nt fragments via Cas3 helicase/nuclease | Creates blunt DSB 3 bp upstream of PAM | Creates staggered DSB with 5' overhangs |
| PAM Requirement | 5'-AAG-3' (on target strand) | 5'-NGG-3' (complementary strand) | 5'-TTTV-3' (target strand) |
| Off-target Rate (with 3 mismatches) | <0.1% | ~2.5% (wild-type) | <0.5% |
The tripartite framework of Adaptation, Expression, and Interference represents a elegantly minimal yet highly effective immune strategy. Research into its origins suggests modular evolution, where components like Cas1 integrases may have originated from ancestral transposons. This staged paradigm provides the direct blueprint for CRISPR-Cas9 technology. Ongoing research into the diversity of these stages across CRISPR types continues to fuel the development of next-generation precision gene-editing tools, antimicrobials, and diagnostics for therapeutic and research applications.
Within the ongoing thesis research into the evolutionary origins of the CRISPR-Cas9 bacterial adaptive immune system, a fundamental understanding of its natural diversity is paramount. This technical guide provides an in-depth overview of the primary classification of CRISPR-Cas systems, which are broadly divided into Class 1 and Class 2. This classification is based on the architecture of their effector modules, a distinction critical for researchers exploring ancestral systems and for professionals engineering novel genetic tools.
Core Classification Principle
CRISPR-Cas systems are universally categorized by the structure of their effector complexes that execute interference (target cleavage). Class 1 systems utilize multi-subunit effector complexes, while Class 2 systems employ a single, large protein for crRNA processing and interference.
Class 1 Systems: Multi-Subunit Effector Complexes
Class 1 systems are the most phylogenetically widespread and are thought to represent the ancestral forms from which Class 2 systems evolved. They are subdivided into Types I, III, and IV.
Type I Systems
- Signature Protein: Cas3, a fused helicase-nuclease.
- Effector Complex: Cascade (CRISPR-associated complex for antiviral defense). A multi-protein complex that binds crRNA, identifies target DNA via PAM recognition, and recruits Cas3 for degradation.
- Subtypes: I-A through I-G.
Type III Systems
- Signature Protein: Cas10, containing HD nuclease and cyclase domains.
- Effector Complex: Csm (Type III-A) or Cmr (Type III-B). Unique for targeting both RNA and DNA transcriptionally coupled to the target RNA. They exhibit collateral cleavage activity.
- Subtypes: III-A, III-B, III-C, III-D.
Type IV Systems
- Signature Protein: Csf1, but often lacking core Cas proteins like Cas3 or Cas10.
- Effector Complex: Multi-subunit. Poorly characterized but implicated in plasmid interference.
- Subtypes: IV-A through IV-C.
Class 2 Systems: Single-Protein Effector Complexes
Class 2 systems are more recently evolved and are the foundation for most genome-engineering applications due to their simplicity. They are subdivided into Types II, V, and VI.
Type II Systems
- Signature Protein: Cas9.
- Mechanism: Uses a single Cas9 protein with RuvC and HNH nuclease domains to cleave both strands of target DNA. Requires tractRNA and RNase III for crRNA maturation.
- Subtypes: II-A, II-B, II-C.
Type V Systems
- Signature Protein: Cas12 (e.g., Cas12a/Cpf1, Cas12b, Cas12f).
- Mechanism: Single-protein effectors with a RuvC-like nuclease domain. Cas12a processes its own pre-crRNA and creates staggered DNA cuts. Many exhibit collateral trans-cleavage of ssDNA.
- Subtypes: V-A through V-K.
Type VI Systems
- Signature Protein: Cas13 (e.g., Cas13a, Cas13b).
- Mechanism: RNA-targeting effectors with two HEPN nuclease domains. Upon binding target RNA, they become promiscuous RNases, leading to collateral RNA cleavage—a property harnessed for diagnostics.
- Subtypes: VI-A through VI-D.
Table 1: Core Characteristics of CRISPR-Cas Classes and Types
| Feature | Class 1 | Class 2 |
|---|---|---|
| Effector Architecture | Multi-subunit complex | Single, multi-domain protein |
| Types | I, III, IV | II, V, VI |
| Representative Proteins | Cas3 (Type I), Cas10 (Type III) | Cas9 (II), Cas12 (V), Cas13 (VI) |
| Pre-crRNA Processing | By dedicated subunit of complex or Cas6 | By the effector itself (II, V) or separate RNase (III) |
| Target Nucleic Acid | DNA (I, IV) / DNA & RNA (III) | DNA (II, V) / RNA (VI) |
| Collateral Activity | Common in Type III | Common in Types V & VI |
| Prevalence in Prokaryotes | ~90% of systems | ~10% of systems |
Table 2: Key Molecular Features of Major Class 2 Effectors
| Effector | Type | PAM Requirement | Cleavage Pattern | Maturation | Collateral Activity? |
|---|---|---|---|---|---|
| Cas9 | II | 3'-NGG (SpCas9) | Blunt-ended DSB | tractRNA + RNase III | No |
| Cas12a | V | 5'-TTTV | Staggered DSB | Self-processing | ssDNA trans-cleavage |
| Cas13a | VI | Protospacer Flanking Site (PFS) | RNA cleavage | Self-processing | ssRNA trans-cleavage |
Detailed Experimental Protocol: Class 2 EffectorIn VitroCharacterization
This protocol is essential for thesis work characterizing novel Cas protein function.
Objective: To reconstitute DNA/RNA cleavage activity of a putative Class 2 effector in vitro and determine its biochemical requirements.
Materials:
- Purified recombinant Cas effector protein.
- Synthetic pre-crRNA and tractRNA (for Type II systems).
- Target DNA plasmid or in vitro-transcribed RNA substrate (fluorescently labeled for quantification).
- Reaction buffer (e.g., 20 mM HEPES-KOH pH 7.5, 100 mM KCl, 5 mM MgCl₂, 1 mM DTT).
- Nuclease-free water, RNase inhibitor (for RNA targets).
- Thermostable incubator.
- Agarose gel electrophoresis or capillary electrophoresis system.
Procedure:
- Ribonucleoprotein (RNP) Complex Formation: In a 1.5 mL tube, combine 100 nM Cas protein with 120 nM crRNA (and 120 nM tractRNA for Cas9) in reaction buffer. Incubate at 37°C for 10 minutes.
- Cleavage Reaction Initiation: Add the target nucleic acid substrate (10 nM) to the RNP complex. Adjust final volume to 20 µL with reaction buffer. Include a negative control without the Cas protein.
- Incubation: Place the reaction mixture in a thermostable incubator at 37°C (or optimal predicted temperature) for 1 hour.
- Reaction Termination: Add 2 µL of Proteinase K (20 mg/mL) and incubate at 56°C for 15 minutes to degrade the Cas protein.
- Product Analysis:
- For DNA Targets: Analyze products by 1% agarose gel electrophoresis. Include DNA ladder. Cleavage yields smaller fragments.
- For RNA Targets: Use denaturing polyacrylamide gel electrophoresis or a capillary electrophoresis bioanalyzer for higher resolution.
- PAM Determination: Repeat the assay using a target plasmid library containing randomized sequences adjacent to the protospacer. Sequence the uncleaved plasmids after negative selection to identify depleted sequences, revealing the PAM.
Visualization of CRISPR-Cas Classification and Function
Title: CRISPR-Cas System Classification Tree and Targets
Title: In Vitro Characterization Workflow for Novel Cas Effectors
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-Cas Classification Research
| Item | Function & Explanation |
|---|---|
| High-Fidelity DNA Polymerase (e.g., Phusion) | For accurate amplification of novel cas gene loci from genomic or metagenomic DNA. |
| Heterologous Expression Vector (e.g., pET series) | Allows for inducible, high-yield expression of Cas proteins in E. coli for purification. |
| Affinity Purification Resin (Ni-NTA or Strep-Tactin) | Enables purification of polyhistidine- or Strep-tag-fused recombinant Cas proteins. |
| In Vitro Transcription Kit (T7) | For generating precise, nuclease-free crRNA, tracrRNA, and target RNA substrates. |
| Fluorescently-Labeled Oligonucleotide Probes | Serve as sensitive targets for cleavage assays; fluorescence allows quantitation of activity and collateral effects. |
| PAM Library Oligo Pool | A synthesized DNA library with randomized bases flanking a constant protospacer sequence, used for empirical PAM determination (SELEX-like assay). |
| RNase Inhibitor (e.g., Recombinant RNasin) | Critical for any experiment involving RNA (Type III, VI systems) to prevent degradation by environmental RNases. |
| Capillary Electrophoresis System (e.g., Bioanalyzer) | Provides high-resolution, quantitative analysis of nucleic acid cleavage products from in vitro assays. |
The dichotomy between Class 1 and Class 2 CRISPR-Cas systems represents a fundamental axis of diversity in this adaptive immune system. For research tracing the evolutionary trajectory from ancestral multi-protein complexes to streamlined single-effect tools, this classification provides the essential framework. The experimental and analytical toolkit continues to evolve, driven by the need to characterize the vast reservoir of unclassified systems in microbial genomes, fueling both basic research into bacterial immunity and the development of next-generation biotechnologies.
This whitepaper details the key historical discoveries that transformed CRISPR-Cas9 from an observation of mysterious genetic repeats into a characterized prokaryotic adaptive immune system. Framed within a broader thesis on CRISPR-Cas9 bacterial immune system origins, this guide provides a technical chronology for research professionals, emphasizing the experimental methodologies that underpinned each breakthrough.
Table 1: Key Historical Milestones in CRISPR-Cas Research
| Year | Discovery/Event | Key Researchers/Group | Primary Experimental Evidence |
|---|---|---|---|
| 1987 | Unusual repeated sequences in E. coli genome reported. | Ishino et al. | Cloning and sequencing of the iap gene region. |
| 2002 | Term "CRISPR" coined; cas genes identified. | Jansen et al. | Bioinformatic analysis of microbial genomes. |
| 2005 | CRISPR spacers derived from foreign genetic elements (viruses, plasmids). | Mojica et al.; Pourcel et al.; Bolotin et al. | Spacer sequence homology to phage/plasmid databases. |
| 2007 | Experimental proof of CRISPR as an adaptive immune system in bacteria. | Barrangou et al. | Phage challenge assays in Streptococcus thermophilus. |
| 2010 | In vitro reconstitution of DNA targeting by Cascade complex. | van der Oost group | Biochemical assays with purified E. coli Cascade and Cas3. |
| 2011 | CRISPR-Cas9 system from Streptococcus pyogenes characterized as a two-RNA-guided DNA endonuclease. | Doudna, Charpentier et al. | In vitro cleavage assays with tracrRNA, crRNA, and Cas9 protein. |
| 2012 | Engineering of dual-RNA into single-guide RNA (sgRNA); programmable DNA cleavage demonstrated. | Doudna, Charpentier et al. | In vitro cleavage of plasmid DNA with chimeric sgRNA. |
Table 2: Quantitative Data from Foundational Experiments
| Experiment (Year) | Critical Quantitative Result | Method of Measurement |
|---|---|---|
| Spacer Analysis (2005) | ~2% of all spacers showed significant homology to known phage/plasmid sequences. | BLASTN alignment against GenBank. |
| Phage Resistance (2007) | Phage-plaque formation reduced by 4 orders of magnitude in CRISPR-Cas+ strains vs. defective mutants. | Plaque assay titer quantification. |
| In vitro Cleavage (2011) | Cas9-mediated plasmid cleavage efficiency of >90% with correct PAM (5'-NGG-3') present. | Gel electrophoresis densitometry. |
Detailed Experimental Protocols
Protocol: Phage Challenge Assay (Barrangou et al., 2007)
Objective: To demonstrate adaptive immunity via CRISPR spacer acquisition. Materials: Streptococcus thermophilus strain, virulent phage, M17 agar plates, phage buffer. Procedure:
- Culture & Infection: Grow phage-sensitive S. thermophilus to mid-log phase. Infect with phage at high MOI (Multiplicity of Infection).
- Recovery & Plating: Allow phage adsorption (10 min), dilute culture, and plate on M17 agar for overnight growth at 37°C.
- Survivor Isolation: Pick surviving bacterial colonies.
- Spacer Analysis: Isolate genomic DNA from survivors and parent strain. Amplify CRISPR locus by PCR, clone, and sequence. Compare spacer arrays.
- Validation of Resistance: Challenge survivors and parent strain with the same phage in a standard plaque assay.
Protocol:In vitroReconstitution of Cas9 DNA Cleavage (Jinek et al., 2012)
Objective: To prove programmable DNA cleavage by Cas9 guided by a chimeric single-guide RNA (sgRNA). Materials: Purified S. pyogenes Cas9 protein, T7 RNA polymerase, DNA oligonucleotides, target plasmid DNA, NTPs, reaction buffer. Procedure:
- sgRNA Synthesis: Transcribe sgRNA in vitro from a dsDNA template containing T7 promoter and sgRNA sequence. Purify via gel electrophoresis or column.
- Cleavage Reaction Assemble:
- 100 nM purified Cas9 protein
- 120 nM sgRNA
- 10 nM target plasmid DNA (containing target site and PAM)
- 20 mM HEPES buffer (pH 7.5), 150 mM KCl, 10 mM MgCl₂, 1 mM DTT, 5% glycerol.
- Incubation: Incubate reaction at 37°C for 60 minutes.
- Analysis: Stop reaction with Proteinase K/EDTA. Analyze cleavage products by agarose gel electrophoresis (0.8% gel). Visualize DNA with ethidium bromide; cleaved linear plasmid runs at a distinct size compared to supercoiled/ nicked circular forms.
Visualizations
Timeline of Key CRISPR Discovery Milestones
Experimental Workflow for Phage Challenge Assay
In vitro Cas9-sgRNA DNA Cleavage Assay Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Foundational CRISPR-Cas Research
| Reagent/Material | Function in Research | Example from Key Studies |
|---|---|---|
| High-Efficiency Competent Cells | For cloning of CRISPR loci and spacer arrays after PCR amplification. | E. coli DH5α or TOP10 cells used in Ishino (1987) and subsequent spacer cloning. |
| Phage Lysate (High Titer) | To provide strong selective pressure in bacterial challenge assays. | Virulent phage for S. thermophilus in Barrangou et al. (2007) experiments. |
| T7 RNA Polymerase Kit | For in vitro transcription of crRNA, tracrRNA, and sgRNA. | Used in Jinek et al. (2011, 2012) to produce guide RNAs for in vitro cleavage. |
| Nickel-NTA Agarose Resin | For purification of His-tagged recombinant Cas9 protein from E. coli expression systems. | Essential for obtaining pure, active Cas9 for biochemical characterization. |
| Target Plasmid with PAM Site | Substrate DNA for in vitro cleavage assays to demonstrate specificity and efficiency. | Custom plasmids containing a target sequence followed by 5'-NGG-3' PAM. |
| Thermostable DNA Polymerase for PCR | To amplify and analyze CRISPR locus architecture from genomic DNA. | Used in all spacer acquisition and diversity studies (e.g., 2005, 2007). |
Harnessing Bacterial Machinery: Engineering CRISPR-Cas9 for Precision Genome Editing
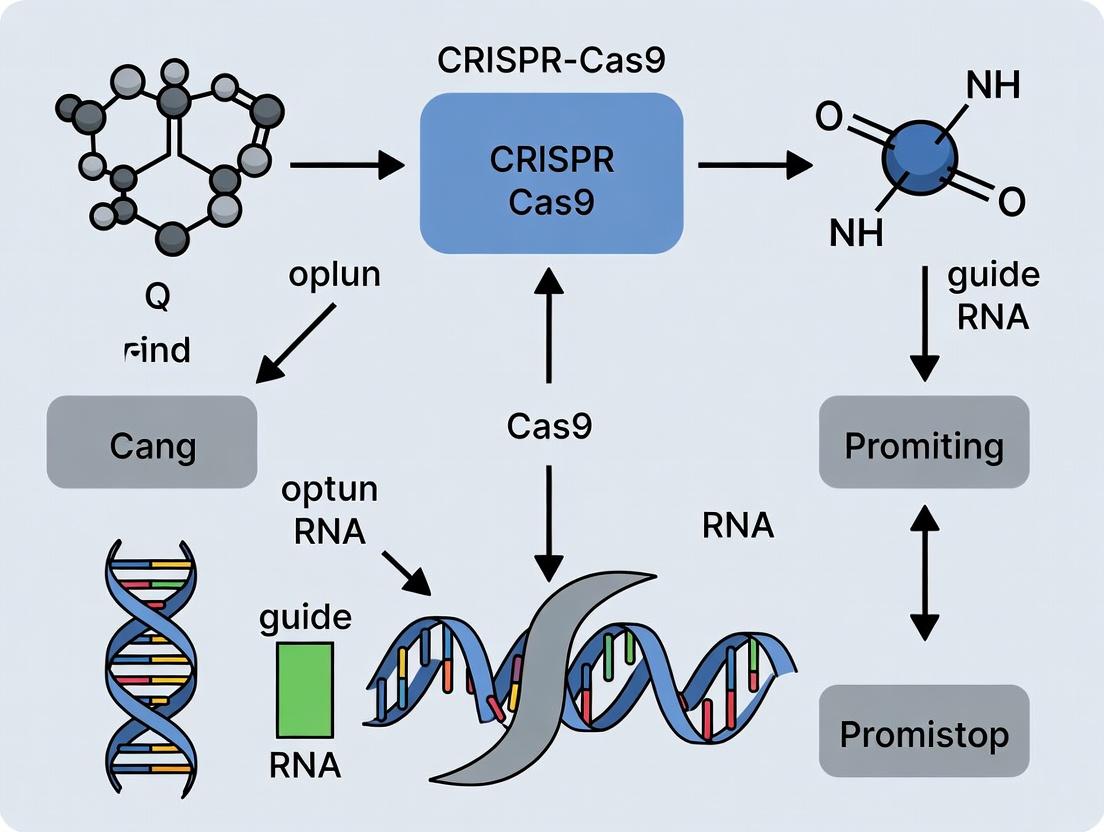
This whitepaper details the fundamental engineering breakthrough that transformed the native CRISPR-Cas9 bacterial immune system into a programmable genome editing tool: the fusion of the dual-RNA guide structure into a single-guide RNA (sgRNA). Framed within research on CRISPR-Cas9's origins as a bacterial adaptive immune system, we explore the structural biology, design principles, and experimental validation of the sgRNA. This adaptation was pivotal in shifting Cas9 from a prokaryotic defense mechanism to a versatile technology for genetic manipulation in eukaryotic cells, revolutionizing molecular biology and therapeutic development.
The type II CRISPR-Cas9 system, derived from Streptococcus pyogenes, provides adaptive immunity in bacteria by utilizing two separate RNA components: the CRISPR RNA (crRNA), which contains a 20-nucleotide spacer sequence complementary to the target DNA, and the trans-activating crRNA (tracrRNA), which base-pairs with the crRNA repeat region and facilitates Cas9 recruitment. This crRNA:tracrRNA duplex, along with the Cas9 endonuclease, forms an RNA-protein complex that surveils and cleaves foreign DNA. The core engineering leap for biomedical application was the rational design of a chimeric single-guide RNA (sgRNA), which combines the essential functional domains of both natural RNAs into a single, programmable molecule.
Structural Basis and Design Rationale
The sgRNA is a synthetic fusion where the 5' end consists of the user-defined ~20 nt guide sequence (replacing the crRNA spacer), followed by a portion of the crRNA repeat sequence, and a linker loop that connects to the tracrRNA-derived sequence. This chimeric RNA maintains the critical secondary structures necessary for Cas9 binding and activation.
Key Structural Domains of sgRNA:
- Guide Sequence (5' end): 18-22 nucleotides defining genomic target via Watson-Crick base pairing.
- CRISPR Repeat-Derived Region: Forms the stem-loop 1, essential for Cas9 recognition.
- Linker Loop: Connects the crRNA- and tracrRNA-derived sequences; length and sequence can affect stability.
- tracrRNA-Derived Sequence: Forms multiple stem-loops (e.g., stem-loop 2 and 3) crucial for Cas9 activation and complex stability.
The following table summarizes the quantitative comparison between the native duplex and the engineered sgRNA.
Table 1: Quantitative Comparison of Native Duplex vs. Engineered sgRNA
| Feature | Native crRNA:tracrRNA Duplex | Engineered Single-Guide RNA (sgRNA) |
|---|---|---|
| Number of RNA Molecules | Two (crRNA ~40 nt, tracrRNA ~89 nt in S. pyogenes) | One (chimeric, typically ~100 nt) |
| Base-Pairing Requirement | Required in trans for complex assembly | Encoded in cis via designed linker |
| Guide Sequence Modification | Requires cloning into CRISPR array | Synthesized as a single oligo or encoded in plasmid |
| Typical Delivery Method in Eukaryotes | Challenging; requires co-expression of both RNAs | Simplified; expression from a single U6 or Pol III promoter |
| Editing Efficiency in Early Validation (Human Cells) | Moderate, dependent on duplex formation | Consistently high, streamlined expression |
| Primary Reference | Deltcheva et al., Nature 2011 | Jinek et al., Science 2012 |
Detailed Experimental Protocol:In VitrosgRNA Validation
The seminal experiment validating sgRNA function (Jinek et al., Science 2012) is outlined below.
A. Materials & Reagents (The Scientist's Toolkit)
- Purified S. pyogenes Cas9 Protein: Recombinant His-tagged Cas9, expressed in E. coli and purified via nickel-affinity chromatography. Function: The endonuclease effector protein.
- DNA Oligonucleotides: Synthetic single-stranded DNA (ssDNA) oligos containing the target sequence (plus PAM, 5'-NGG-3') and a non-target control. Function: Substrates for in vitro cleavage assays.
- T7 RNA Polymerase Kit: For in vitro transcription (IVT) of sgRNA from a DNA template. Function: Generates high yields of sgRNA.
- PCR System: To generate dsDNA targets from cloned plasmid or overlapping oligos. Function: Provides dsDNA substrates for cleavage.
- Reaction Buffer (NEBuffer 3.1): Provides optimal ionic strength (100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, pH 7.9) for Cas9 nuclease activity. Function: Supports enzymatic cleavage.
- Proteinase K & RNAse A: For reaction termination and digestion of Cas9/sgRNA. Function: Cleans up samples for gel analysis.
- Polyacrylamide or Agarose Gel Electrophoresis System: For separation and visualization of cleaved vs. uncleaved DNA fragments.
B. Step-by-Step Methodology
- sgRNA Synthesis: Design a DNA template with a T7 promoter upstream of the sgRNA sequence. Perform IVT using the T7 RNA polymerase kit. Purify the sgRNA via phenol-chloroform extraction and ethanol precipitation. Resuspend in RNase-free water and quantify.
- Target DNA Preparation: Anneal complementary ssDNA oligos to form a short (~100-200 bp) dsDNA target containing the PAM site, or amplify a target region from a plasmid via PCR. Purify the dsDNA product.
- Cas9 RNP Complex Assembly: In a 1.5 mL tube, combine:
- Purified Cas9 protein (final conc. ~100 nM)
- In vitro transcribed sgRNA (final conc. ~120 nM)
- 1X Reaction Buffer
- Incubate at 37°C for 10 minutes to allow ribonucleoprotein (RNP) complex formation.
- Cleavage Reaction: Add target dsDNA (final conc. ~10 nM) to the pre-assembled RNP complex. Incubate at 37°C for 1 hour.
- Reaction Termination: Add Proteinase K (to degrade Cas9) and optionally RNase A (to degrade sgRNA). Incubate at 56°C for 15 minutes.
- Analysis: Load the samples onto an agarose or polyacrylamide gel. Include controls: DNA only, DNA + Cas9 (no guide), DNA + sgRNA (no Cas9). Visualize via ethidium bromide or SYBR Safe staining. Successful cleavage is indicated by the appearance of two lower molecular weight bands corresponding to the predicted fragment sizes.
Impact on Eukaryotic Genome Editing Workflow
The sgRNA format drastically simplified the delivery and expression of the CRISPR-Cas9 system in mammalian cells. The workflow transition is illustrated below.
Diagram 1: From Native Bacterial Immunity to Engineered Eukaryotic Tool
Key Research Reagent Solutions
Table 2: Essential Toolkit for sgRNA-Based CRISPR-Cas9 Research
| Reagent/Material | Function & Role in sgRNA Context |
|---|---|
| sgRNA Expression Vector (e.g., pX330 derivative) | Plasmid containing a U6 promoter driving sgRNA transcription and a CBh promoter driving Cas9. Allows stable delivery and expression of both components from a single plasmid. |
| Synthetic sgRNA (chemically modified) | For RNP delivery. High-purity, IVT or chemically synthesized sgRNA, often with 2'-O-methyl modifications at terminals to enhance stability and reduce immunogenicity. |
| Cas9 Protein (purified) | For in vitro assays or RNP delivery. Recombinant Cas9, often with nuclear localization signals (NLS) for eukaryotic use, complexed with sgRNA to form active editing complexes. |
| Custom dsDNA or ssDNA Oligos | Serve as templates for sgRNA in vitro transcription, or as homology-directed repair (HDR) donors for precise editing alongside the sgRNA/Cas9 system. |
| NLS-Peptide Conjugates | Used to non-covalently complex with sgRNA:Cas9 RNP to enhance nuclear import in certain delivery strategies (e.g., electroporation). |
| Lipid Nanoparticles (LNPs) | A key delivery vehicle for therapeutic sgRNA/Cas9 RNPs or mRNA/sgRNA combinations, encapsulating them for efficient in vivo delivery to target tissues. |
The creation of the sgRNA was not merely a simplification but a core re-engineering of a bacterial immune component. It resolved the critical bottleneck of co-delivering and processing two separate RNAs in eukaryotic cells, making CRISPR-Cas9 accessible, efficient, and programmable. This leap, grounded in understanding the original biological function, enabled the transition from basic research on microbial immunity to a platform technology with profound implications for functional genomics, cellular engineering, and the development of next-generation genetic therapies. Ongoing research continues to optimize sgRNA chemistry, structure, and delivery, further expanding the capabilities of this foundational technology.
Design Principles for Target Selection and sgRNA Construction
The CRISPR-Cas9 system, repurposed from a prokaryotic adaptive immune system, has revolutionized genetic engineering. Understanding its origins—where archaea and bacteria capture spacers from invasive genetic elements to direct Cas nucleases for cleavage—is fundamental to its applied use. This guide details the core design principles for selecting target sequences and constructing single guide RNAs (sgRNAs) that underpin effective gene editing, framed by insights from this ancestral immune function. Precision here is paramount, mirroring the specificity required for the system to distinguish self from non-self in its native context.
Core Design Principles for Target Selection
Effective CRISPR editing begins with the selection of an optimal target sequence within the genomic DNA. This process mirrors the spacer acquisition phase in bacterial immunity, where specificity and avoidance of self-targeting are critical for survival.
Sequence Characteristics
- Protospacer Adjacent Motif (PAM): The target site must be adjacent to a PAM sequence specific to the Cas nuclease used. For the commonly used Streptococcus pyogenes Cas9 (SpCas9), the PAM is 5'-NGG-3', located immediately downstream (3') of the target sequence on the non-complementary strand.
- Target Sequence (Protospacer): Typically 20 nucleotides immediately 5' to the PAM. It must be unique in the genome to minimize off-target effects.
- GC Content: Optimal GC content is between 40-60%. This promotes stable sgRNA-DNA binding without excessive rigidity.
- Avoidance of Homopolymer Runs: Sequences with stretches of 4 or more identical nucleotides (e.g., AAAA, CCCC) should be avoided as they can impair editing efficiency.
- Genomic Context: Target sites should be within an accessible chromatin region. Sites in open, transcriptionally active chromatin (euchromatin) are generally more efficient than those in condensed, silent regions (heterochromatin).
Off-Target Assessment
A primary challenge is avoiding cleavage at genomic loci with high sequence similarity to the intended target. Computational tools must be used to scan the reference genome for potential off-target sites with up to 3-5 mismatches, particularly in the "seed" region proximal to the PAM (positions 1-12). High-fidelity Cas9 variants (e.g., SpCas9-HF1, eSpCas9) can be employed to mitigate this risk.
Table 1: Quantitative Parameters for Optimal Target Selection
| Parameter | Optimal Range/Value | Rationale |
|---|---|---|
| Protospacer Length | 20 nt | Standard length for SpCas9; balances specificity and efficiency. |
| PAM Sequence (SpCas9) | 5'-NGG-3' | Absolute requirement for SpCas9 recognition and cleavage. |
| GC Content | 40% - 60% | Ensures sufficient binding energy and secondary structure avoidance. |
| Distance from DSB | < 10 bp from intended edit | Editing efficiency (HDR) decreases with distance from the double-strand break (DSB). |
| Off-Target Mismatch Tolerance (Seed) | 0 mismatches in seed region (positions 1-12) | Mismatches in the seed region severely reduce or abolish cleavage. |
| Predicted On-Target Score (e.g., from CRISPOR) | > 60 | Composite score predicting high cleavage activity. |
Experimental Protocol: In Silico Target Site Identification and Validation
Methodology:
- Define Genomic Locus: Identify the exact chromosomal coordinates of the gene or regulatory element of interest using a reference genome (e.g., GRCh38/hg38).
- PAM Scanning: Use software (e.g., CRISPOR, Benchling, CHOPCHOP) to scan both DNA strands for all instances of the appropriate PAM sequence within your locus.
- Extract Candidate Protospacers: For each PAM, extract the 20 nucleotides directly 5' to it.
- Filter and Rank: Apply filters for GC content, absence of homopolymers, and predicted secondary structure of the sgRNA. The software will generate specificity scores (e.g., Doench '16 efficiency score, MIT specificity score) for ranking.
- Off-Target Analysis: For the top 3-5 candidates, run a genome-wide off-target search. Prioritize candidates with zero or minimal predicted off-target sites, especially those with few mismatches in the seed region.
- Final Selection: Select at least 2-3 sgRNAs per target for empirical validation, as predictive algorithms are not infallible.
Design Principles for sgRNA Construction
The sgRNA is a chimeric RNA that replaces the native CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA) of the bacterial system. Its design dictates the specificity and efficiency of DNA cleavage.
sgRNA Scaffold and Expression
The sgRNA consists of two primary components:
- Target-Specific CrRNA Sequence: The 5' 20-nucleotide sequence that is complementary to the genomic target.
- Structural Scaffold (tracrRNA): A conserved ~80 nt sequence that forms a duplex with the crRNA and binds to Cas9, facilitating its cleavage activity.
sgRNAs are typically expressed from RNA Polymerase III promoters (e.g., U6, H1) in mammalian cells to ensure precise 5' and 3' ends. For bacterial work, T7 promoters are common.
Table 2: Key Design Considerations for sgRNA Expression Constructs
| Component | Design Principle | Function |
|---|---|---|
| Promoter | U6 (human), 7SK, H1, or T7 (in vitro/bacterial) | Drives high-level, Pol III-dependent expression with precise transcription start. |
| Target Sequence | Cloned directly downstream of promoter. Must match genomic target (excluding PAM). | Provides sequence specificity for DNA recognition. |
| sgRNA Scaffold | Conserved sequence downstream of target. Must be correctly folded. | Binds Cas9 protein and facilitates DNA cleavage. |
| Terminator | 4-6 Thymidines (T) for Pol III; self-cleaving ribozyme for Pol II. | Signals transcription termination. Poly-T tract is the simplest terminator for U6. |
Experimental Protocol: Cloning sgRNA into an Expression Vector
Methodology (Golden Gate Assembly Example):
- Oligonucleotide Design: Design forward and reverse oligonucleotides (ultramers) encoding your 20-nt target sequence with 4-5 bp overhangs compatible with your chosen cloning site (e.g., BbsI for Addgene's pSpCas9(BB) backbone).
- Annealing: Resuspend oligonucleotides to 100 µM. Mix 1 µL of each, 1 µL of 10x T4 Ligase Buffer, and 7 µL nuclease-free water. Heat to 95°C for 5 minutes, then ramp cool to 25°C over 45 minutes.
- Golden Gate Assembly: Set up a reaction with 50 ng of linearized backbone vector, 1 µL of the diluted (1:100) annealed oligo duplex, 1 µL of BbsI (Type IIs restriction enzyme), 1 µL of T7 DNA Ligase, and 2 µL of 10x T4 Ligase Buffer. Bring to 20 µL with water.
- Cycling: Perform a thermocycler program: (37°C for 5 min, 20°C for 5 min) x 30 cycles, then 80°C for 10 min.
- Transformation: Transform 2-5 µL of the assembly reaction into competent E. coli, plate on selective antibiotic, and incubate overnight.
- Validation: Pick colonies, culture, and purify plasmid DNA. Validate by Sanger sequencing using a primer that binds upstream of the sgRNA insert.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-Cas9 Target Validation
| Reagent / Material | Function | Example Product/Provider |
|---|---|---|
| High-Fidelity Cas9 Nuclease | Engineered variant with reduced off-target cleavage. | Alt-R S.p. HiFi Cas9 Nuclease V3 (IDT) |
| Chemically Modified sgRNA | Synthetic sgRNA with phosphorothioate bonds and 2'-O-methyl analogs; increases stability and reduces immune response. | Synthego sgRNA EZ Kit |
| T7 Endonuclease I (T7EI) | Enzyme to detect mismatches in heteroduplex DNA formed after NHEJ repair; used for initial cleavage efficiency assessment. | New England Biolabs T7 Endonuclease I |
| Next-Generation Sequencing (NGS) Library Prep Kit for CRISPR | Enables deep sequencing of target loci to quantify editing efficiency and profile off-target events. | Illumina CRISPR Amplicon Sequencing Kit |
| Guide-it Genotype Confirmation Kit | A PCR-based assay for detecting indels via fragment length analysis. | Takara Bio Guide-it Genotype Confirmation Kit |
| GEN1- 1 HDR Enhancer | Small molecule that improves Homology-Directed Repair (HDR) efficiency for precise edits. | (Available from various chemical suppliers) |
| Control sgRNA (Non-Targeting) | A sgRNA with no perfect match in the host genome; essential for controlling for non-specific effects of transfection and Cas9 activity. | Scrambled Control sgRNA (Santa Cruz Biotechnology) |
Visualizations
Diagram 1: CRISPR-Cas9 Targeting & Bacterial Immunity Analogy
Diagram 2: sgRNA Design & Cloning Workflow
Diagram 3: Key sgRNA Expression Vector Components
The CRISPR-Cas system, derived from an adaptive bacterial immune defense against invading bacteriophages, has revolutionized genetic engineering. The translation of this bacterial mechanism into eukaryotic cells, however, hinges entirely on the development of efficient, safe delivery vehicles. This whitepaper details the core delivery technologies enabling CRISPR-Cas9 clinical translation, tracing their conceptual lineage from prokaryotic transformation to human therapeutic vectors.
Prokaryotic Origins: Bacterial Transformation & Electroporation
The foundational delivery method, bacterial transformation, allows for plasmid introduction. A refined, high-efficiency version is electroporation, critical for CRISPR research.
Experimental Protocol: Bacterial Electroporation for CRISPR Plasmid Transformation
- Competent Cell Preparation: Grow E. coli strain (e.g., DH5α) to mid-log phase (OD600 ≈ 0.5-0.6). Chill culture on ice.
- Cell Washing: Pellet cells by centrifugation (4°C, 4000xg, 10 min). Gently resuspend in ice-cold, sterile 10% glycerol solution. Repeat wash 2-3 times.
- Electroporation: Mix 50 μL competent cells with 1-10 ng plasmid DNA (e.g., CRISPR-Cas9 expression vector) in pre-chilled electroporation cuvette (1mm gap). Apply electrical pulse (typical settings: 1.8 kV, 200 Ω, 25 μF). Immediately add 1 mL SOC recovery medium.
- Recovery & Selection: Incubate with shaking at 37°C for 1 hour. Plate onto LB agar containing appropriate antibiotic (e.g., ampicillin, 100 μg/mL). Incubate overnight at 37°C.
- Validation: Isolve plasmid from resulting colonies via miniprep and confirm by restriction digest and sequencing.
Quantitative Data: Transformation Efficiency
| Transformation Method | Typical Efficiency (CFU/μg DNA) | Key Parameter | Optimal DNA Type/Size |
|---|---|---|---|
| Chemical Competence | 1 x 10⁷ – 1 x 10⁸ | Heat-Shock (42°C) | Plasmid DNA (<15 kb) |
| Electroporation | 1 x 10⁹ – 3 x 10¹⁰ | Field Strength (12-15 kV/cm) | Plasmid DNA, Linear Fragments |
Viral Vectors: From Phage Biology to Clinical Gene Therapy
Adeno-Associated Viruses (AAVs) and Lentiviruses (LVs) are the primary viral vectors for in vivo and ex vivo CRISPR delivery, respectively.
Experimental Protocol: Production of VSV-G Pseudotyped Lentivirus for CRISPR Delivery
- Vector & Packaging Plasmids: Co-transfect HEK293T cells (at 70-80% confluency in 10cm dish) with:
- Transfer plasmid (e.g., lentiCRISPRv2): 10 μg
- Packaging plasmid (psPAX2): 7.5 μg
- Envelope plasmid (pMD2.G): 2.5 μg Use a transfection reagent like PEI (Polyethylenimine, 1 mg/mL, 60 μL).
- Media Change: Replace media 6-8 hours post-transfection with fresh DMEM + 10% FBS.
- Harvest: Collect virus-containing supernatant at 48 and 72 hours post-transfection. Pool harvests.
- Concentration: Filter supernatant (0.45 μm), then concentrate via ultracentrifugation (70,000xg, 2h, 4°C) or using PEG-it virus precipitation solution.
- Titration: Transduce HEK293 cells with serial dilutions of vector. Perform qPCR for integrated vector genome or assay for antibiotic resistance (e.g., puromycin) to determine titer (TU/mL).
Quantitative Data: Clinical Viral Vectors
| Vector | Packaging Capacity | Tropism | Integration | Typical In Vivo Titer (vg/mL) | Key Advantage | Major Safety Concern |
|---|---|---|---|---|---|---|
| AAV | ~4.7 kb | Broad (serotype-dependent) | No (episomal) | 1 x 10¹³ – 1 x 10¹⁴ | Low immunogenicity, Long-term expression | Pre-existing immunity, Capsid toxicity |
| Lentivirus | ~8 kb | Broad (pseudotype-dependent) | Yes (random) | 1 x 10⁸ – 1 x 10⁹ (transducing units) | High efficiency, Large cargo capacity | Insertional mutagenesis |
Non-Viral Vectors: Lipid Nanoparticles (LNPs)
LNPs have emerged as the leading non-viral platform for systemic CRISPR-Cas9 mRNA/sgRNA delivery, exemplified by the clinical success of patisiran and COVID-19 mRNA vaccines.
Experimental Protocol: Microfluidic Formulation of CRISPR-LNPs
- Lipid Stock Preparation: Dissolve ionizable lipid (e.g., DLin-MC3-DMA), cholesterol, DSPC, and PEG-lipid (e.g., DMG-PEG2000) in ethanol at molar ratio 50:38.5:10:1.5.
- Aqueous Phase Preparation: Dilute CRISPR-Cas9 mRNA and sgRNA in citrate buffer (pH 4.0) at a total RNA concentration of 0.2 mg/mL.
- Mixing: Use a microfluidic mixer (e.g., NanoAssemblr Ignite). Set flow rate ratio (aqueous:organic) to 3:1, with a total combined flow rate of 12 mL/min.
- Dialysis & Formulation: Immediately dilute the formed LNPs in PBS (pH 7.4). Dialyze against PBS for 24h at 4°C to remove ethanol and establish neutral pH.
- Characterization: Measure particle size and PDI by dynamic light scattering (DLS, target: 70-100 nm, PDI <0.2). Determine encapsulation efficiency using RiboGreen assay.
Quantitative Data: LNP Formulation Components & Performance
| LNP Component | Example Compound | Molar Ratio (%) | Primary Function |
|---|---|---|---|
| Ionizable Cationic Lipid | DLin-MC3-DMA, SM-102 | 50 | Binds nucleic acid, promotes endosomal escape |
| Cholesterol | Cholesterol | 38.5 | Stabilizes bilayer structure |
| Helper Phospholipid | DSPC | 10 | Improves bilayer stability and fusogenicity |
| PEGylated Lipid | DMG-PEG2000 | 1.5 | Controls particle size, reduces aggregation, shields surface |
Physical Methods: Electroporation forEx VivoDelivery
Clinical ex vivo CRISPR editing (e.g., for CAR-T cells or hematopoietic stem cells) relies heavily on nucleofection, an advanced electroporation technique.
Experimental Protocol: Nucleofection of Primary Human T Cells with CRISPR RNP
- Cell Preparation: Isolate CD3+ T cells from PBMCs using magnetic beads. Activate with CD3/CD28 antibodies for 24-48 hours.
- RNP Complex Formation: Mix recombinant S. pyogenes Cas9 protein (30 pmol) with synthetic sgRNA (60 pmol, targeting TRAC locus) in PBS. Incubate at room temp for 10 min.
- Nucleofection: Wash 1-2 x 10⁶ T cells, resuspend in 100 μL of specified nucleofection solution (e.g., P3 Primary Cell Kit). Add pre-formed RNP complex and transfer to cuvette. Run program (e.g., EO-115 on 4D-Nucleofector).
- Recovery & Expansion: Immediately add pre-warmed medium + IL-7/IL-15 to cuvette. Transfer cells to culture plate. Expand for 7-14 days.
- Analysis: Assess editing efficiency by T7E1 assay or NGS. Validate phenotype by flow cytometry for CD3.
The Scientist's Toolkit: Research Reagent Solutions
| Reagent/Material | Supplier Examples | Function in Delivery/Editing Workflow |
|---|---|---|
| LentiCRISPRv2 Plasmid | Addgene | All-in-one lentiviral vector for expression of Cas9, sgRNA, and puromycin resistance. |
| Recombinant S. pyogenes Cas9 Protein | Thermo Fisher, IDT | For rapid formation of RNP complexes for electroporation; reduces off-target effects. |
| Lipofectamine CRISPRMAX | Thermo Fisher | A lipid-based transfection reagent optimized for delivery of CRISPR RNPs or plasmids to difficult cell lines. |
| Neon Transfection System | Thermo Fisher | Electroporation system for high-efficiency transfection of CRISPR components into mammalian cells. |
| AAVpro Purification Kit | Takara Bio | For purification and concentration of high-titer, high-purity AAV vectors from cell lysates. |
| P3 Primary Cell 4D-Nucleofector Kit | Lonza | Optimized reagents for nucleofection of hard-to-transfect primary cells like T cells and HSCs. |
| Ribogreen RNA Quantitation Kit | Thermo Fisher | Assay for accurately determining RNA encapsulation efficiency within LNPs. |
| T7 Endonuclease I | NEB | Enzyme for detecting CRISPR-induced indels via mismatch cleavage (surveyor assay). |
Technical Visualizations
Title: Bacterial CRISPR Origin to Delivery Tool Evolution (77 chars)
Title: LNP Formulation and Delivery Workflow (44 chars)
Title: Lentiviral Vector Production Pipeline (45 chars)
The revolutionary genome engineering tools in use today are direct intellectual and technological derivatives of the adaptive immune system found in bacteria and archaea. The core thesis of their origin posits that the CRISPR-Cas9 system evolved as a mechanism for prokaryotes to record and destroy invasive genetic elements, such as bacteriophages and plasmids. This natural function—DNA sequence recognition and cleavage by the Cas9 nuclease guided by a CRISPR RNA (crRNA)—has been repurposed. The paradigms of knockout, knock-in, base editing, and transcriptional regulation represent the logical extension of this bacterial defense apparatus, moving from destroying invading DNA to precisely editing, regulating, or rewriting genomic information in eukaryotic cells.
Gene Knockout via Non-Homologous End Joining (NHEJ)
Principle: This paradigm most closely mimics the native function of the bacterial immune system: creating a double-strand break (DSB) in target DNA. In eukaryotic cells, the error-prone NHEJ repair pathway often introduces small insertions or deletions (indels) during repair, leading to frameshift mutations and gene disruption.
Detailed Protocol for Mammalian Cell Knockout:
- Design & Cloning: Design a 20-nt guide RNA (gRNA) sequence complementary to an early exon of the target gene. Clone this sequence into a plasmid expressing the gRNA scaffold and a Cas9 nuclease (e.g., Streptococcus pyogenes SpCas9).
- Delivery: Transfect the plasmid into target cells (e.g., HEK293T) using a suitable method (lipofection, electroporation).
- Selection & Expansion: Apply appropriate antibiotics (e.g., puromycin) 24-48 hours post-transfection to select for transfected cells. Culture for 5-7 days to allow for gene editing and protein turnover.
- Validation: Harvest genomic DNA. Perform PCR amplification of the target locus. Analyze indels via T7 Endonuclease I assay or next-generation sequencing (NGS).
Key Quantitative Data on Knockout Efficiency:
| Parameter | Typical Range | Notes |
|---|---|---|
| Indel Formation Efficiency | 20-80% | Highly dependent on cell type, gRNA design, and delivery efficiency. |
| NHEJ Repair Fidelity | Error-prone (~65% of DSBs) | Precise repair without indels occurs in ~35% of cases. |
| Common Indel Size | 1-10 bp | Larger deletions (>50 bp) possible but less frequent. |
Diagram: Workflow for CRISPR-Cas9 Mediated Gene Knockout
Gene Knock-in via Homology-Directed Repair (HDR)
Principle: Exploits the alternative, high-fidelity HDR pathway. Co-delivery of a donor DNA template with homology arms flanking the DSB site allows for precise insertion of exogenous sequences (e.g., fluorescent tags, SNPs).
Detailed Protocol for Precise Knock-in:
- Design Components: Design gRNA to cut near the desired integration site. Synthesize a donor template (single-stranded oligodeoxynucleotide - ssODN or double-stranded DNA - dsDNA) containing the desired edit flanked by homology arms (70-100 nt for ssODN, >500 bp for dsDNA).
- Synchronization: For dividing cells, synchronize to S/G2 phase where HDR is more active. Use small molecule inhibitors (e.g., Scr7 to suppress NHEJ, RS-1 to enhance HDR).
- Co-delivery: Co-electroporate Cas9 ribonucleoprotein (RNP) complex and the donor template.
- Screening: Enrich edited cells via FACS (if knock-in includes a fluorescent marker) or antibiotic selection. Screen clones by PCR and sequencing to confirm precise integration.
Key Quantitative Data on Knock-in Efficiency:
| Parameter | Typical Range | Notes |
|---|---|---|
| HDR Efficiency (ssODN) | 1-20% | Efficiency drops sharply with larger inserts. |
| HDR vs. NHEJ Ratio | ~1:10 to 1:50 | NHEJ is dominant in most mammalian cells. |
| Optimal Homology Arm Length | 70-100 nt (ssODN) | Longer arms (>500 bp) for dsDNA templates. |
Base Editing
Principle: Evolved from Cas9 to achieve direct, irreversible chemical conversion of one base pair to another without creating a DSB or requiring a donor template. Fusion of a catalytically impaired Cas9 (nickase) to a deaminase enzyme enables direct C•G to T•A (Cytosine Base Editors - CBEs) or A•T to G•C (Adenine Base Editors - ABEs) conversion.
Detailed Protocol for Single-Nucleotide Conversion:
- Editor Selection: Choose appropriate base editor (e.g., BE4max for C-to-T, ABE8e for A-to-G) based on desired change and sequence context (must be within the editing window, typically protospacer positions 4-8).
- gRNA Design: Design gRNA to position the target base within the editing window. The PAM must be present, but no DSB will occur.
- Delivery: Transfect base editor plasmid or deliver as RNP.
- Analysis: Harvest genomic DNA 3-7 days post-transfection. Amplify target region by PCR and analyze via Sanger sequencing or high-throughput sequencing to quantify editing efficiency and purity (minimizing indels).
Diagram: Mechanism of a Cytosine Base Editor (CBE)
Transcriptional Regulation (CRISPRa/i)
Principle: Derived from the concept of catalytically dead Cas9 (dCas9), which binds DNA without cutting. Fusion of transcriptional effector domains (e.g., VP64, p65, KRAB) to dCas9 allows for targeted gene activation (CRISPRa) or repression (CRISPRi), mimicking prokaryotic regulatory networks but with programmability.
Detailed Protocol for Gene Activation (CRISPRa):
- System Assembly: Use a dCas9-VP64-p65-Rta (VPR) activator fusion protein. Design gRNAs to target the promoter or enhancer region of the gene of interest, typically within -200 to +1 bp relative to the transcription start site (TSS).
- Delivery: Co-transfect dCas9-VPR and gRNA expression plasmids into cells.
- Stable Line Generation: For sustained regulation, generate stable cell lines via lentiviral transduction of dCas9-effector and gRNA.
- Validation: Measure mRNA levels via qRT-PCR 48-72 hours post-transfection/induction. Assess protein levels via western blot or immunofluorescence.
Key Quantitative Data on Transcriptional Regulation:
| Parameter | Typical Range (CRISPRa) | Typical Range (CRISPRi) |
|---|---|---|
| Activation Fold-Change | 10x - 1000x+ | N/A |
| Repression Efficiency | N/A | 50% - 90% reduction |
| Key Targeting Region | -200 to +1 bp from TSS | -50 to +300 bp from TSS |
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent/Material | Function & Purpose |
|---|---|
| SpCas9 Nuclease (WT & D10A) | Wild-type for DSB creation; D10A nickase mutant for base editing or reduced off-targets. |
| dCas9 (Catalytically Dead Cas9) | DNA-binding platform for transcriptional regulation, epigenome editing, and imaging. |
| Base Editor Plasmids (BE4, ABE8e) | All-in-one expression vectors for efficient C-to-T or A-to-G conversion. |
| Chemically Modified sgRNA | Synthetic gRNAs with 2'-O-methyl and phosphorothioate modifications enhance stability and RNP activity. |
| HDR Donor Templates (ssODN) | Single-stranded DNA oligos for precise point mutations and small tag insertions via HDR. |
| NHEJ Inhibitors (e.g., Scr7) | Small molecules to temporarily suppress NHEJ, improving HDR efficiency in dividing cells. |
| Lentiviral dCas9-Effector Particles | For stable, inducible, and efficient delivery of transcriptional regulators to diverse cell types. |
| T7 Endonuclease I / Surveyor Nuclease | Enzymes for initial detection and quantification of indel mutations post-knockout. |
| Next-Generation Sequencing Kits | For comprehensive, quantitative analysis of editing outcomes (indels, base edits, HDR). |
Diagram: Comparison of Core CRISPR Application Paradigms
The journey from a fundamental study of how bacteria fend off viruses to the suite of precision genome engineering tools outlined here epitomizes transformative basic research. Each paradigm—knockout, knock-in, base editing, and transcriptional regulation—solves a distinct biological or therapeutic problem by creatively modifying the core components of the CRISPR-Cas system. Understanding their operational details, efficiencies, and limitations, as framed by their prokaryotic origins, empowers researchers to select and implement the optimal strategy for their specific experimental or therapeutic goals.
The discovery of the CRISPR-Cas9 system as an adaptive immune mechanism in bacteria has revolutionized biological research. Originating from the study of Streptococcus pyogenes and other prokaryotes, this system provides a memory of past viral infections, enabling sequence-specific targeting and cleavage of foreign genetic material. This whitepaper frames the application of CRISPR libraries for high-throughput screening (HTS) within the context of this foundational thesis: understanding the bacterial immune origins of CRISPR-Cas9 is not merely an academic exercise but is critical for optimizing its precision, efficiency, and safety as a screening tool. Modern CRISPR screening libraries are direct technological descendants of this natural defense system, repurposed for systematic functional genomics in mammalian cells to identify genes involved in specific phenotypes, from essential genes for survival to novel drug targets for oncology and infectious disease.
Core Principles of CRISPR Screening Libraries
CRISPR libraries are pooled collections of lentiviral vectors, each encoding a single-guide RNA (sgRNA) designed to knock out (using Cas9 nuclease) or modulate (using dCas9 fused to transcriptional activators/repressors) a specific gene. In a typical genome-wide screen, tens of thousands of cells are transduced at a low multiplicity of infection (MOI) to ensure one sgRNA per cell, creating a complex, representative knockout pool.
Key Library Types:
- Genome-wide Knockout Libraries: Target every protein-coding gene (e.g., 18,000+ genes) with multiple sgRNAs per gene (e.g., 4-10) for robustness. Popular examples include the Brunello (human) and Brie (mouse) libraries.
- Focused/Sublibraries: Target a specific gene family (e.g., kinases, GPCRs) or pathway.
- CRISPRa (Activation): Use dCas9-VPR to overexpress genes from their native promoters.
- CRISPRi (Interference): Use dCas9-KRAB to transcriptionally repress genes.
Recent data from leading providers (e.g., Addgene, Horizon Discovery) and publications highlight the standardization and scale of available resources.
Table 1: Comparative Overview of Common Genome-wide CRISPR Knockout Libraries
| Library Name | Species | Target Genes | sgRNAs/Gene | Total sgRNAs | Core Application | Reference (PMID) |
|---|---|---|---|---|---|---|
| Brunello | Human | 19,114 | 4 | 76,456 | High-confidence knockout; reduced off-target | 26780180 |
| Toronto KnockOut (TKO) v3 | Human | 18,053 | 4 | 70,948 | Identification of essential genes | 26780180 |
| Mouse Brie | Mouse | 20,611 | 4 | 82,444 | Genome-wide screening in murine cells | 29601079 |
| GeCKO v2 | Human/Mouse | 19,050 (Human) | 3-6 per gene | 123,411 (total) | Dual-species; versatile knockout | 23287718 |
| CRISPRa v2 (SAM) | Human | 23,430 | 3-5 | 70,290+ | Transcriptional activation | 28067908 |
Detailed Experimental Protocol for a Positive Selection Survival Screen
The following protocol outlines a standard positive selection screen to identify genes essential for cell proliferation or survival under a specific condition (e.g., drug treatment).
A. Screen Design & Library Amplification
- Library Selection: Choose an appropriate library (e.g., Brunello for human cells).
- Plasmid Amplification: Transform the library plasmid pool into electrocompetent E. coli and plate on large-format LB-ampicillin agar plates. Scrape and maxiprep the pooled bacteria to obtain high-diversity plasmid DNA. Titre the library by sequencing to confirm sgRNA representation.
B. Lentiviral Production & Cell Line Engineering
- Day 1: Seed HEK293T cells in a 10cm dish.
- Day 2: Transfert using polyethylenimine (PEI) with three plasmids:
- Library sgRNA plasmid (e.g., lentiCRISPRv2): 10 µg
- Packaging plasmid (psPAX2): 7.5 µg
- Envelope plasmid (pMD2.G): 2.5 µg
- Day 3 & 4: Replace medium. Harvest viral supernatant at 48h and 72h post-transfection, filter (0.45 µm), and concentrate using PEG-it virus precipitation solution.
- Titration: Transduce target cells (e.g., A549) with serial dilutions of virus plus polybrene (8 µg/ml). Select with puromycin (1-3 µg/ml, determined by kill curve) for 7 days. Calculate viral titer (TU/ml) based on percentage of surviving cells.
C. Large-Scale Screen Transduction & Selection
- Transduction: Seed 2e7 target cells. Transduce at an MOI of ~0.3 to ensure most cells receive ≤1 sgRNA, maintaining >500x library coverage (e.g., for a 76k sgRNA library, use ~4e7 transduced cells). Include polybrene.
- Selection: 24h post-transduction, add puromycin. Maintain selection for 5-7 days until all cells in an untransduced control dish are dead.
D. Phenotypic Selection & Sample Collection
- Day 0 Sample (T0): Harvest ~1e7 cells (covering library >100x) as a genomic DNA (gDNA) baseline.
- Phenotype Application: Split the remaining population into experimental (e.g., +Drug) and control (DMSO) arms. Passage cells for 14-21 population doublings under selective pressure.
- Endpoint Samples (T_end): Harvest >1e7 cells from each arm.
E. Next-Generation Sequencing (NGS) & Analysis
- gDNA Extraction: Use a silica-membrane based kit for all samples (T0, Tendcontrol, Tendtreated).
- sgRNA Amplification: Perform a two-step PCR.
- PCR1: Amplify the sgRNA region from gDNA (5-10 µg per reaction, multiple reactions per sample) using indexed primers complementary to the lentiviral backbone.
- PCR2: Add Illumina adapters and sample-specific barcodes.
- Sequencing: Pool purified PCR products and sequence on an Illumina NextSeq (75bp single-end).
- Bioinformatic Analysis:
- Align reads to the library sgRNA reference file.
- Count reads per sgRNA for each sample.
- Normalize counts and use algorithms (e.g., MAGeCK, CERES) to compare sgRNA abundance between T_end and T0, or between treatment and control. This identifies sgRNAs (and thus genes) that are significantly depleted (essential genes) or enriched (resistance genes).
Title: CRISPR Screening Workflow: Library to Analysis
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for CRISPR Screening
| Item | Function & Critical Notes | Example Product/Supplier |
|---|---|---|
| Validated CRISPR Library | Pre-designed, sequence-verified pooled sgRNA plasmid library. Ensures even representation and target efficacy. | Brunello Human CRISPR Knockout Pooled Library (Addgene #73179) |
| Lentiviral Packaging Plasmids | Required for safe production of replication-incompetent viral particles. psPAX2 (gag/pol) and pMD2.G (VSV-G envelope) are standard. | psPAX2 (Addgene #12260), pMD2.G (Addgene #12259) |
| Stable Cas9-Expressing Cell Line | Cells constitutively expressing Cas9 nuclease. Simplifies screen to delivery of sgRNA library only. | A549-Cas9, HEK293T-Cas9 (commercially available) |
| Polyethylenimine (PEI) | High-efficiency, low-cost cationic polymer for transfection of packaging cells. | Linear PEI, MW 25,000 (Polysciences) |
| Polybrene (Hexadimethrine bromide) | A cationic polymer that enhances viral transduction efficiency by neutralizing charge repulsion. | Polybrene (Sigma-Aldrich TR-1003) |
| Puromycin Dihydrochloride | Selection antibiotic for cells transduced with puromycin-resistance containing vectors (e.g., lentiCRISPRv2). | Puromycin (Gibco A1113803) |
| Next-Generation Sequencing Kit | For preparing amplified sgRNA libraries for sequencing on Illumina platforms. | Illumina DNA Prep Kit |
| sgRNA Read Counting & Analysis Software | Computational tool for quantifying sgRNA depletion/enrichment and identifying hit genes. | MAGeCK (open source), Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) |
Pathway & Hit Validation
Primary screen hits require rigorous validation. This involves a multi-step process to exclude false positives and confirm phenotype causality.
Title: CRISPR Screen Hit Validation Cascade
Validation Protocol: Clonal Cell Line Generation
- Design Validation sgRNAs: Design 2-3 new sgRNAs distinct from the screening library, targeting the hit gene.
- Clone into Lentiviral Vector: Clone individual sgRNAs into a lentiviral CRISPR vector (e.g., lentiCRISPRv2).
- Generate Polyclonal and Clonal Pools: Transduce target cells, select with puromycin. For clonal lines, perform limiting dilution in 96-well plates to obtain single-cell colonies.
- Verify Knockout: For each clone, confirm gene disruption via:
- Genomic DNA Sequencing: PCR amplify the target region, sequence, and analyze for indels.
- Western Blot: Confirm loss of protein expression (if antibody available).
- Phenotype Re-test: Subject validated knockout clones and a non-targeting sgRNA control clone to the original screening condition (e.g., drug treatment). Measure phenotype (e.g., viability, apoptosis, reporter signal) to confirm the initial observation.
High-throughput screening with CRISPR libraries represents the operationalization of a fundamental bacterial immune principle for systematic mammalian functional genomics. By leveraging the programmable, DNA-targeting specificity derived from the CRISPR-Cas9 system's origins, researchers can now conduct unprecedentedly precise and scalable genetic screens. This guide outlines the core technical and practical considerations for executing such screens, from library selection and viral production to NGS analysis and multi-layered validation. As the field evolves, linking these powerful screening methodologies back to the core biology of Cas protein diversity and mechanism—continuing the thesis of its bacterial origins—will be key to developing next-generation screening tools with enhanced specificity, novel functionalities, and broader therapeutic applications.
The revolutionary application of CRISPR-Cas9 in modern therapeutics is a direct descendant of fundamental research into prokaryotic adaptive immunity. Our broader thesis on the origins of the CRISPR-Cas bacterial immune system reveals that nature's solution for phage defense—characterized by sequence-specific targeting, memory, and cleavage—has been exapted to create two dominant therapeutic paradigms. This guide details the ex vivo and in vivo strategies constituting the current pipeline, grounded in the mechanistic principles derived from its bacterial ancestry.
Ex Vivo Therapeutic Strategies
Ex vivo therapy involves genetic modification of a patient's cells outside the body, followed by reinfusion.
Key Applications & Clinical Data
Table 1: Select Ex Vivo CRISPR Therapies in Clinical Development (2023-2024)
| Therapeutic Product (Company/Institution) | Target Gene / Cell Type | Indication | Clinical Phase | Key Efficacy Metric (Latest Data) | | :--- | :--- | : --- | :--- | :--- | | CTX001 (Vertex/CRISPR Tx) | BCL11A in hematopoietic stem/progenitor cells (HSPCs) | β-Thalassemia, Sickle Cell Disease | Approved (US, UK, EU) | 94% of β-thal patients (n=48) transfusion-independent; 100% of SCD patients (n=44) free of severe vaso-occlusive crises (≥12mo follow-up) | | OTQ923/HIX763 (Novartis) | BCL11A in HSPCs | Sickle Cell Disease | Phase I/II | Mean fetal hemoglobin 26.7% at 3 months post-infusion (n=7) | | GPH101 (Graphite Bio) | Corrects β-globin gene in HSPCs | Sickle Cell Disease | Phase I/II (Halted) | Insertion efficiency >40% in preclinical models | | EDIT-301 (Editas Medicine) | Erythroid enhancer of BCL11A in HSPCs | Sickle Cell Disease, β-Thalassemia | Phase I/II | Sustained fetal hemoglobin >40% in first SCD patient at 6 months | | CAR-T cells (Various) | PD-1 or TCR genes in T-cells | Oncology (Solid Tumors) | Phase I/II | Varied; one study showed 30% objective response rate in NSCLC with PD-1 KO CAR-T |
Detailed Ex Vivo Protocol: CRISPR Editing of HSPCs for Sickle Cell Disease
This protocol is derived from clinical trial methodologies for BCL11A-targeting therapies.
Objective: Generate CRISPR-Cas9 edited CD34+ hematopoietic stem and progenitor cells (HSPCs) for autologous transplantation.
Materials (Research Reagent Solutions):
- Mobilized Leukapheresis Product: Source of patient CD34+ HSPCs.
- CD34+ Cell Selection Kit (e.g., CliniMACS): For positive immunomagnetic selection of target cells.
- Stem Cell Growth Medium (e.g., StemSpan SFEM II): Serum-free medium optimized for HSPC expansion.
- CRISPR RNP Complex: Comprising:
- S.p. Cas9 Nuclease (GMP-grade): 100 µg/mL final reaction concentration.
- Synthetic sgRNA (targeting BCL11A erythroid enhancer): 120 µg/mL final concentration. Pre-complexed at 37°C for 10 minutes.
- Electroporation System (e.g., Lonza 4D-Nucleofector): For RNP delivery.
- Ancillary Reagents: Recombinant human cytokines (SCF, TPO, FLT3-L), Cas9 ELISA kit, NGS-based off-target assay panel.
Workflow:
- Cell Harvest & Selection: Isolate mononuclear cells from leukapheresis. Enrich CD34+ cells via immunomagnetic selection. Assess viability (must be >95%) and purity (must be >90%).
- Pre-stimulation: Culture selected CD34+ cells at 1-2x10^6 cells/mL in growth medium with cytokines (100ng/mL SCF, 100ng/mL TPO, 100ng/mL FLT3-L) for 24-48 hours at 37°C, 5% CO2.
- CRISPR RNP Electroporation: Harvest pre-stimulated cells. Resuspend at 1x10^8 cells/mL in electroporation buffer. Mix 100µL cell suspension with 10µL pre-complexed RNP. Electroporate using designated pulse code (e.g., EO-115 on 4D-Nucleofector). Immediately transfer to pre-warmed culture medium.
- Post-editing Culture & QC: Culture cells for 48-72 hours. Sample for:
- Editing Efficiency: T7E1 assay or NGS of target locus (goal: >60% indels).
- Viability & Yield: Trypan blue exclusion.
- Sterility, Mycoplasma, Endotoxin: GMP-release testing.
- Cryopreservation & Infusion: Cryopreserve edited cells in 10% DMSO. After myeloablative conditioning (e.g., busulfan), thaw and infuse cells intravenously into the patient.
Ex Vivo Cell Therapy Manufacturing Workflow
In Vivo Therapeutic Strategies
In vivo therapy involves direct administration of genetic medicines to the patient to edit cells within the body.
Key Applications & Clinical Data
Table 2: Select In Vivo CRISPR Therapies in Clinical Development (2023-2024)
| Therapeutic Product (Company) | Delivery System | Target Gene / Tissue | Indication | Clinical Phase | Key Efficacy/Safety Metric |
|---|---|---|---|---|---|
| NTLA-2001 (Intellia/Regeneron) | LNP | TTR in hepatocytes | Transthyretin Amyloidosis | Phase III | Serum TTR reduction: 93% mean at 28 days (1mg/kg dose). 0.8% mild infusion-related reactions. |
| VRTX-B (Vertex/CRISPR Tx) | LNP | Unknown in hepatocytes | Hereditary Angioedema | Phase I/II | >90% reduction in kallikrein activity reported. |
| NTLA-2002 (Intellia) | LNP | KLKB1 in hepatocytes | Hereditary Angioedema | Phase II | 95% mean reduction in kallikrein at highest dose. Well-tolerated. |
| EDIT-101 (Editas/Allergan) | AAV5 (subretinal) | CEP290 in photoreceptors | Leber Congenital Amaurosis 10 | Phase I/II | 3 of 14 patients showed measurable BCVA improvement. No serious ocular events. |
| KB407 (Krystal Biotech) | HSV-1 Vector | CFTR in airway epithelium | Cystic Fibrosis | Phase I | Preclinical: restored 50% of CFTR function in human air-liquid interface models. |
Detailed In Vivo Protocol: Systemic LNP Delivery for Liver-Targeted Editing
This protocol is modeled on clinical-stage programs like NTLA-2001 for TTR amyloidosis.
Objective: Formulate and administer CRISPR-Cas9 mRNA and sgRNA via lipid nanoparticles (LNPs) for targeted gene disruption in hepatocytes.
Materials (Research Reagent Solutions):
- Ionizable Cationic Lipid (e.g., DLin-MC3-DMA or proprietary variants): Key component for encapsulation and endosomal escape.
- Helper Lipids: Cholesterol, DSPC, PEGylated lipid (e.g., DMG-PEG2000). Stabilize particle structure and control pharmacokinetics.
- CRISPR Payload: In vitro transcribed or synthetic:
- Cas9 mRNA (Pseudouridine-modified): 0.5 mg/mL in citrate buffer.
- sgRNA (targeting gene of interest): 0.5 mg/mL in citrate buffer.
- Microfluidic Mixer (e.g., NanoAssemblr): For precise, reproducible LNP formation.
- Tangential Flow Filtration (TFF) System: For buffer exchange and concentration of formed LNPs.
- Analytical Tools: Dynamic Light Scattering (DLS) for size, RiboGreen assay for encapsulation efficiency, HPLC for lipid composition.
Workflow:
- LNP Formulation (Rapid Mixing): Prepare an ethanol phase containing ionizable lipid, cholesterol, DSPC, and PEG-lipid at molar ratio (e.g., 50:38.5:10:1.5). Prepare an aqueous phase containing Cas9 mRNA and sgRNA at equimolar ratio in citrate buffer (pH 4.0). Use a microfluidic mixer to combine the two phases at a fixed flow rate ratio (typically 3:1 aqueous:ethanol). The change in pH causes self-assembly into LNPs.
- Buffer Exchange & Concentration: Immediately dilute the crude LNP mixture in PBS (pH 7.4). Concentrate and dialyze against PBS using TFF to remove ethanol and establish neutral pH.
- Characterization: Measure particle size (target: 70-100 nm, PDI <0.2) via DLS. Determine RNA encapsulation efficiency (>90% target) via fluorescent dye assay. Confirm sterility and endotoxin levels.
- In Vivo Administration: Administer via bolus intravenous injection in animal models or human patients at a defined dose (e.g., 1.0 mg RNA per kg body weight). Monitor for acute reactions.
- Pharmacodynamic Analysis: Collect serial blood and tissue biopsies. Assess target protein reduction (e.g., serum TTR via ELISA) and gene editing in liver DNA via NGS.
LNP Formulation for In Vivo CRISPR Delivery
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for CRISPR Therapeutic Pipeline Research
| Reagent / Material | Function in Research & Development | Example Use Case |
|---|---|---|
| High-Fidelity Cas9 Variants (e.g., HiFi Cas9, eSpCas9) | Reduces off-target editing while maintaining high on-target activity. | Critical for ex vivo editing of HSPCs to minimize genotoxic risk. |
| Base Editor (BE) & Prime Editor (PE) Plasmids/mRNA | Enables precise point mutations or small insertions without double-strand breaks. | Developing therapies for SNPs (e.g., APOE4, PKU) where silencing is not desired. |
| GMP-grade sgRNA Synthesis Kits | Production of clinical-grade, highly pure, endotoxin-free guide RNA. | Scale-up manufacturing for both ex vivo and in vivo therapeutics. |
| LNP Formulation Screening Kits | Allows rapid empirical testing of ionizable lipid libraries for optimal in vivo delivery. | Identifying novel LNP formulations for targeting tissues beyond liver (e.g., lung, CNS). |
| All-in-One NGS Off-Target Analysis Panel | Comprehensive genome-wide detection of potential off-target sites. | Mandatory safety assessment for IND-enabling studies of any CRISPR therapeutic. |
| Immunodeficient Mouse Models (e.g., NSG) | Supports engraftment and study of human xenografts for ex vivo edited cells. | Preclinical efficacy and toxicology testing of engineered HSPCs or CAR-T cells. |
| AAV Serotype Library (AAV9, AAVPHP.eB, AAV-DJ) | Enables tropism testing for in vivo delivery to specific tissues (CNS, muscle, eye). | Developing gene editing therapies for neurological or muscular disorders. |
Beyond the Cut: Solving Specificity, Efficiency, and Delivery Challenges in CRISPR-Cas9 Systems
The CRISPR-Cas9 system, derived from a bacterial adaptive immune defense mechanism, has revolutionized genome engineering. Its precision, however, is not absolute. The "off-target problem"—the cleavage of DNA sites with sequence homology to the intended guide RNA (gRNA)—poses a significant risk for therapeutic applications and functional genomics. This guide details the molecular mechanisms underlying off-target effects and elaborates on two seminal, high-sensitivity detection methods, framed within the evolutionary context of the CRISPR-Cas system's origins in prokaryotic immunity.
Mechanisms of Off-Target Cleavage
The Cas9 endonuclease, guided by a single RNA (sgRNA), identifies target DNA via protospacer adjacent motif (PAM) recognition and RNA-DNA complementarity. Off-target events occur when Cas9 tolerates mismatches, bulges, or non-canonical PAMs. Mechanistically, this tolerance is rooted in the energetics of R-loop formation; sufficient base-pairing stability, even with imperfections, can trigger nuclease activation. This imperfect fidelity may reflect the system's evolutionary origin in bacteria, where a robust defense against rapidly evolving phages required balancing specificity with the ability to recognize related viral strains.
Detection Methodologies
GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing)
GUIDE-seq detects double-strand breaks (DSBs) in situ by capturing integration events of a blunt, double-stranded oligodeoxynucleotide (dsODN) tag.
Detailed Protocol:
- Transfection: Co-deliver Cas9-sgRNA ribonucleoprotein (RNP) complexes and the GUIDE-seq dsODN tag (typically 34-36 bp, phosphorothioate-modified) into target cells.
- Tag Integration: Endogenous repair pathways, primarily non-homologous end joining (NHEJ), incorporate the dsODN tag at DSB sites.
- Genomic DNA Extraction & Shearing: Harvest cells 48-72 hours post-transfection. Extract and sonicate genomic DNA to ~500 bp fragments.
- Library Preparation: Perform end-repair, A-tailing, and ligation of sequencing adapters. Use biotinylated primers specific to the dsODN tag for selective PCR enrichment of tag-containing fragments.
- Sequencing & Analysis: Perform paired-end sequencing. Map reads to the reference genome, identify tag integration sites, and calculate read counts. Peaks indicate DSB loci. Off-target sites are identified by comparison to the on-target sequence.
Quantitative Data Summary: Table 1: Typical GUIDE-seq Performance Metrics
| Metric | Value/Range | Notes |
|---|---|---|
| Detection Sensitivity | Down to ~0.1% of on-target reads | Can identify low-frequency events |
| Required Sequencing Depth | 30-50 million reads (human genome) | Depth scales with genome complexity |
| Background Noise | Very Low | Due to specific tag integration |
| Time from transfection to data | 7-10 days | Includes cell culture, library prep, and sequencing |
CIRCLE-seq (Circularization forIn VitroReporting of Cleavage Effects by Sequencing)
CIRCLE-seq is an in vitro, highly sensitive method that uses circularized genomic DNA as a substrate for Cas9 cleavage, drastically reducing background signal.
Detailed Protocol:
- Genomic DNA Isolation & Fragmentation: Extract genomic DNA from target cells and fragment it (e.g., via sonication) to ~300 bp.
- Circularization: Use a high-fidelity DNA ligase to circularize the fragmented DNA under dilute conditions to promote intramolecular ligation.
- Linearization & Phosphorylation: Digest circles with Cas9-sgRNA RNP in vitro. Simultaneously, a restriction enzyme (e.g., MmeI) cleaves within a constant adapter sequence, linearizing only circles that were not cut by Cas9. T4 PNK phosphorylates the 5' ends.
- Adapter Ligation & PCR: Ligate sequencing adapters to the MmeI-generated ends, which now flank any Cas9 cut site. Amplify via PCR.
- Sequencing & Analysis: Sequence and map reads. Breakpoints align precisely with Cas9 cleavage sites. Computational filtering removes residual background.
Quantitative Data Summary: Table 2: Typical CIRCLE-seq Performance Metrics
| Metric | Value/Range | Notes |
|---|---|---|
| Detection Sensitivity | Extremely High (can detect <0.01% activity) | In vitro setup minimizes background |
| Required Sequencing Depth | 10-20 million reads | Less complex library than cellular methods |
| Background Noise | Extremely Low | Cleavage background enzymatically removed |
| Experimental Timeline | 3-5 days | No cell culture required |
Visualizing Workflows and Relationships
CRISPR-Cas9 Action and Cellular Repair Pathways
GUIDE-seq Experimental Workflow
CIRCLE-seq Experimental Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Off-Target Detection Studies
| Reagent/Material | Function & Role in Experiment |
|---|---|
| Recombinant Cas9 Nuclease | High-purity, endotoxin-free protein for forming RNP complexes, ensuring consistent activity in GUIDE-seq transfection or in vitro CIRCLE-seq cleavage. |
| Chemically Modified GUIDE-seq dsODN | Blunt-ended, phosphorothioate-protected double-stranded oligo serving as the NHEJ-integrated tag for DSB capture. Modifications prevent degradation. |
| High-Fidelity DNA Ligase (e.g., Circligase) | Critical for CIRCLE-seq to efficiently form circular DNA templates from fragmented gDNA, minimizing concatemers. |
| T4 Polynucleotide Kinase (PNK) | Phosphorylates 5' ends after MmeI digestion in CIRCLE-seq, enabling subsequent adapter ligation for sequencing library construction. |
| Biotinylated PCR Primers (GUIDE-seq) | Enable streptavidin-bead based enrichment of DNA fragments containing the integrated dsODN tag, drastically reducing background for sequencing. |
| MmeI Type IIS Restriction Enzyme | Precisely linearizes uncut circular DNA in CIRCLE-seq, creating a defined breakpoint for adapter ligation and suppressing non-specific background. |
| Next-Generation Sequencing Kit (Illumina-compatible) | For preparing high-complexity sequencing libraries from enriched or amplified DNA fragments for genome-wide analysis. |
The CRISPR-Cas9 system, a cornerstone of modern genome engineering, is a direct descendant of adaptive immune mechanisms in bacteria and archaea. In its native context, the system provides defense against mobile genetic elements (MGEs) like phages and plasmids through DNA capture, crRNA biogenesis, and target interference. A critical feature of this primitive immune system is fidelity—the necessity to discriminate between self (the bacterial genome, housed within CRISPR arrays) and non-self (invasive DNA). This evolutionary pressure to avoid autoimmunity resulted in natural mechanisms like PAM recognition, seed sequence binding, and conformational proofreading. Our engineering of CRISPR-Cas9 for precise mammalian genome editing mirrors this ancient requirement: minimizing off-target cleavage while maintaining robust on-target activity is paramount for research and therapeutic applications. This whitepaper details modern strategies—inspired by and advancing beyond natural evolution—to achieve this goal through protein engineering and guide RNA design.
High-Fidelity Cas9 Variants: Mechanisms and Quantitative Comparison
High-fidelity (Hi-Fi) Cas9 variants are engineered to reduce non-specific DNA interactions, often by destabilizing the RuvC nuclease domain's engagement with DNA or by altering DNA binding kinetics to favor perfectly matched sequences.
Table 1: Comparison of High-Fidelity Streptococcus pyogenes Cas9 (SpCas9) Variants
| Variant Name | Key Mutations (Relative to Wild-Type SpCas9) | Reported Reduction in Off-Target Activity (vs. WT) | On-Target Efficiency (Relative to WT) | Primary Engineering Strategy | Key Reference |
|---|---|---|---|---|---|
| SpCas9-HF1 | N497A, R661A, Q695A, Q926A | >85% reduction across validated sites | Comparable at most sites | Weakening hydrogen bonding to DNA phosphate backbone | Kleinstiver et al., Nature, 2016 |
| eSpCas9(1.1) | K848A, K1003A, R1060A | >90% reduction across validated sites | Comparable at most sites | Altering positive charge to reduce non-specific DNA interactions | Slaymaker et al., Science, 2016 |
| HypaCas9 | N692A, M694A, Q695A, H698A | ~70-90% reduction | Comparable or slightly reduced | Stabilizing the REC3 domain to favor proofreading | Chen et al., Nature, 2017 |
| evoCas9 | M495V, Y515N, K526E, R661Q | Undetectable by GUIDE-seq at 4/5 sites | 30-70% of WT, context-dependent | Phage-assisted continuous evolution (PACE) | Casini et al., Nature Biotech, 2018 |
| SuperFi-Cas9 | R691A | ~3,000-fold reduction for mismatches at positions 18-20 | ~10-50x slower on-target cleavage in vitro; cellular efficiency comparable to SpCas9-HF1 | Prevents conformational activation on mismatched targets | Brakke et al., Science, 2022 |
Table 2: Fidelity Comparison of Cas9 Orthologs & Engineered Variants
| Nuclease | PAM Requirement | Size (aa) | Relative Fidelity (Theoretical) | Relative On-Target Efficiency | Best Suited For |
|---|---|---|---|---|---|
| Wild-Type SpCas9 | NGG | 1368 | Low (Baseline) | High | Initial screens, non-therapeutic models |
| SpCas9-HF1/eSpCas9 | NGG | 1368 | High | High (General) | Most in vitro and in vivo research applications |
| evoCas9 | NGG | 1368 | Very High | Moderate | Applications where utmost specificity is critical |
| SaCas9 | NNGRRT | 1053 | Higher than SpCas9 (kinetically slower) | Moderate | AAV delivery for in vivo gene therapy |
| Cas12a (Cpfl) | T-rich (TTTV) | ~1300 | High (produces staggered ends) | Variable (target-dependent) | Multiplexed editing, AT-rich regions |
Diagram 1: Mechanistic Basis of High-Fidelity Cas9 Variants
Engineered sgRNAs for Enhanced Specificity
The sgRNA structure can be modified to modulate Cas9 binding kinetics and specificity.
Table 3: Engineered sgRNA Scaffold and Truncation Strategies
| Strategy | Design | Proposed Mechanism | Effect on Fidelity | Effect On-Target Potency |
|---|---|---|---|---|
| Truncated sgRNAs (tru-gRNAs) | 17-18nt spacer instead of 20nt | Shortened seed region reduces lifetime of mismatched complexes | Up to 5,000-fold reduction in some off-targets | Can be significantly reduced |
| Extended sgRNAs (e-sgRNAs) | 20nt spacer + 5' GG or GGG extension | 5' guanines enhance seed region stability | 10-100 fold reduction | Generally maintained or slightly improved |
| Chemically Modified sgRNAs | 2'-O-methyl, phosphorothioate at 3' terminus | Increased nuclease resistance, alters kinetics | Moderate improvement (context-dependent) | Improved cellular stability/potency |
| Splinted sgRNAs | Separate crRNA and tracrRNA with partial complementarity | May allow better regulation of complex assembly | Under investigation | Variable |
Diagram 2: sgRNA Engineering Strategies Workflow
Experimental Protocol: GUIDE-seq for Comprehensive Off-Target Profiling
Objective: Genome-wide identification of CRISPR-Cas9 nuclease off-target sites.
Materials:
- Target cells (e.g., HEK293T, primary T-cells)
- Cas9 protein (WT and Hi-Fi variant) + sgRNA complex or expression plasmids
- GUIDE-seq Oligos: Double-stranded, phosphorothioate-modified oligodeoxynucleotides (PTO-dsODNs).
- Transfection reagent (e.g., Lipofectamine CRISPRMAX)
- Genomic DNA extraction kit
- PCR reagents, NEBNext Ultra II DNA Library Prep Kit
- NGS platform (Illumina)
Procedure:
- Complex Formation & Delivery: Co-deliver Cas9-sgRNA ribonucleoprotein (RNP) and 100-200 nM PTO-dsODN into cells via nucleofection/transfection.
- Genomic Integration: The PTO-dsODN integrates into double-strand breaks (DSBs) created by Cas9, both on- and off-target.
- Genomic DNA Harvest: 72 hours post-delivery, extract gDNA.
- Library Preparation: a. Shear & Size-Select: Fragment gDNA to ~500 bp. b. End-Repair, A-tailing, Adapter Ligation: Perform using standard NGS library prep kits. c. GUIDE-seq Amplicon Enrichment: Perform two nested PCRs using: - Outer Primers: One primer specific to the integrated PTO-dsODN, one primer specific to the NGS adapter. - Inner Primers (Indexed): Add sample-specific barcodes and full NGS adapter sequences.
- Sequencing & Analysis: Pool libraries and sequence on a MiSeq or HiSeq. Analyze using the GUIDE-seq analysis pipeline (e.g., from
https://github.com/aryeelab/guideseq) to map PTO-dsODN integration sites as off-target loci.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 4: Key Reagent Solutions for High-Fidelity CRISPR Research
| Item | Function & Description | Example Vendor/Catalog |
|---|---|---|
| High-Fidelity Cas9 Expression Plasmids | Mammalian expression vectors for SpCas9-HF1, eSpCas9, HypaCas9, etc. | Addgene (various, e.g., #72247 for SpCas9-HF1) |
| Purified Hi-Fi Cas9 Nuclease (WT controls) | Recombinant protein for RNP delivery, ensuring controlled stoichiometry. | Integrated DNA Technologies (IDT), Thermo Fisher Scientific |
| Chemically Modified Synthetic sgRNAs | Alt-R CRISPR-Cas9 sgRNAs with 2'-O-methyl and phosphorothioate modifications for stability. | Integrated DNA Technologies (IDT) |
| GUIDE-seq PTO Oligo Duplex | Double-stranded tag for capturing DSBs in off-target profiling assays. | Integrated DNA Technologies (IDT, Alt-R GUIDE-seq Kit) |
| CIRCLE-seq Kit | In vitro method for comprehensive, amplification-based off-target discovery. | IDT (Alt-R CIRCLE-seq Kit) |
| Next-Generation Sequencing Library Prep Kit | For preparing amplicons from GUIDE-seq, CIRCLE-seq, or targeted deep sequencing. | Illumina (Nextera XT), NEB (Ultra II) |
| Off-Target Analysis Software | In silico prediction and NGS data analysis tools. | Benchling (CRISPR guides), CRISPOR, CRISPResso2, GUIDE-seq pipeline |
| Nucleofection System | For high-efficiency delivery of RNPs into difficult cell types (e.g., primary cells). | Lonza (4D-Nucleofector) |
| T7 Endonuclease I (T7E1) or Surveyor Assay | Quick, gel-based method for initial on-target editing and large indel detection. | NEB (M0302L) / IDT |
| Amplicon-EZ NGS Service | Service for targeted deep sequencing of on-target and predicted off-target loci. | Genewiz, Azenta |
The study of CRISPR-Cas9 as a genome engineering tool is inextricably linked to its origins as a bacterial adaptive immune system. Our broader thesis research investigates the evolutionary pressures that shaped the Streptococcus pyogenes Cas9 (SpCas9) system, focusing on how protospacer-adjacent motif (PAM) recognition and spacer acquisition mechanisms optimize defense against bacteriophages. This evolutionary optimization for efficiency and specificity in a native chromatin-free prokaryotic environment creates a fundamental challenge when repurposing the system for eukaryotic genome editing. The core principles of sgRNA design—rooted in bacterial spacer selection—must now be reconciled with the complex landscape of eukaryotic chromatin. This whitepaper synthesizes current design rules with chromatin accessibility data to provide a framework for maximizing editing efficiency in therapeutic and research applications.
Core sgRNA Design Rules: Quantitative Principles
Effective sgRNA design balances on-target efficiency with off-target minimization. The following rules are derived from large-scale pooled screens and biochemical studies.
Table 1: Quantitative Parameters for On-Target sgRNA Design Efficiency
| Parameter | Optimal Feature / Value | Impact on Efficiency (Relative) | Rationale & Biological Origin |
|---|---|---|---|
| GC Content | 40-60% | High (+30-50%) | Stabilizes RNA-DNA heteroduplex; mirrors stable prokaryotic spacers. |
| Position-Specific Nucleotides | Guanine at position 20 (last 5'), 'G' or 'C' at position 1 | High (+20-40%) | Enhances RNA Polymerase III transcription initiation (for U6 promoters) and R-loop stability. |
| Thermodynamic Stability | Low ΔG at PAM-distal end, High ΔG at PAM-proximal end | Moderate (+15-25%) | Facilitates R-loop initiation and propagation; analogous to Cas9 interrogation kinetics in bacteria. |
| sgRNA Length | 20-nt spacer (standard) | Baseline | Matches the spacer length acquired in the native bacterial immune response. |
| PAM Sequence (SpCas9) | NGG (canonical), NAG (alternate) | NGG: High; NAG: Low (~4x less) | Directly inherited from the bacterial system's requirement for precise viral DNA recognition. |
Table 2: Off-Target Sensitivity Predictors
| Predictor | High-Risk Indicator | Mitigation Strategy |
|---|---|---|
| Seed Region Mismatches | ≥1 mismatch in PAM-proximal 10-12 nt | Avoid targets with homologous seed regions elsewhere in genome. |
| Overall Homology | >14-nt matches with 1-3 mismatches elsewhere | Use truncated sgRNAs (17-18 nt) for increased specificity, albeit with potential efficiency trade-off. |
| Genomic Context | Repetitive elements, paralogous genes | Perform rigorous in silico off-target scanning (e.g., Cas-OFFinder). |
The Chromatin Accessibility Challenge
In eukaryotes, nucleosome occupancy and histone modifications create a physical and chemical barrier absent in bacteria. This significantly modulates Cas9 binding and cleavage kinetics.
Table 3: Impact of Chromatin Features on Editing Efficiency
| Chromatin Feature | Effect on Cas9 Efficiency | Supporting Data (Approx. Fold Change) | Proposed Mechanism |
|---|---|---|---|
| Open Chromatin (DNase I Hypersensitive Sites) | Increase | +2 to +5 fold | Enhanced Cas9 DNA binding and R-loop formation. |
| Active Histone Marks (H3K4me3, H3K27ac) | Increase | +1.5 to +3 fold | Recruitment of chromatin remodelers, looser DNA compaction. |
| Repressive Histone Marks (H3K9me3, H3K27me3) | Decrease | -3 to -10 fold | Steric hindrance from nucleosomes; condensed heterochromatin. |
| Nucleosome Occupancy | Decrease (if over target) | -5 to -20 fold | Physical blockade of PAM/spacer sequence accessibility. |
Integrated Experimental Protocol: sgRNA Design with Chromatin Profiling
This protocol outlines a comprehensive workflow for designing and testing sgRNAs informed by chromatin accessibility.
Protocol 4.1: In Silico Design and Prioritization
- Target Identification: Define genomic coordinate of desired edit.
- PAM Scanning: Identify all NGG (and optional NAG) sites within a 50bp window of the target site.
- Chromatin Data Integration:
- Obtain ATAC-seq, DNase-seq, or MNase-seq data for your specific cell type.
- Align candidate sgRNA target sites with chromatin accessibility peaks. Prioritize targets within open chromatin regions.
- Cross-reference with histone modification ChIP-seq data (e.g., from ENCODE). Favor targets co-localized with H3K4me3.
- sgRNA Scoring:
- Apply algorithm-based scoring (e.g., from Doench et al., 2016 Nature Biotechnology).
- Integrate chromatin accessibility score as a weighted multiplier (e.g., 1.5x for open, 0.5x for closed).
- Perform stringent off-target analysis using tools like Cas-OFFinder.
- Final Selection: Select 3-5 top-ranked sgRNAs per target for empirical validation.
Protocol 4.2: Empirical Validation via T7 Endonuclease I (T7EI) Assay
- Purpose: To measure actual editing efficiency of designed sgRNAs in your cellular system.
- Materials: Designed sgRNA expression plasmids (e.g., U6-driven), Cas9 expression construct, transfection reagent, target cells, genomic DNA extraction kit, PCR reagents, T7 Endonuclease I enzyme.
- Procedure:
- Co-transfection: Deliver Cas9 and individual sgRNA constructs into target cells.
- Harvest Genomic DNA: Extract gDNA 72 hours post-transfection.
- PCR Amplification: Amplify a ~500-800bp region flanking the target site.
- Heteroduplex Formation: Denature and reanneal PCR products to allow mismatches from indels.
- T7EI Digestion: Digest heteroduplex DNA with T7EI, which cleaves at mismatch sites.
- Gel Electrophoresis: Analyze products on agarose gel. Cleaved bands indicate successful editing.
- Efficiency Quantification: Use band intensity to calculate indel percentage: % Indel = 100 × (1 - √(1 - (b+c)/(a+b+c))), where a is undigested product, and b & c are cleavage products.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents and Tools for sgRNA Optimization
| Item | Function & Relevance to Design Rules | Example Product/Resource |
|---|---|---|
| Chromatin Accessibility Data | Cell-type-specific maps (ATAC-seq/DNase-seq) for target site prioritization. | ENCODE Consortium database; cell-line-specific datasets from GEO. |
| sgRNA Design Algorithms | Integrates sequence features into a predictive efficiency score. | Broad Institute's "CRISPR Design Tool" (score from Doench et al.), CHOPCHOP. |
| Off-Target Prediction Tool | Identifies potential off-target sites for specificity assessment. | Cas-OFFinder, COSMID. |
| Validated Cas9 Expression System | Consistent, high-activity Cas9 delivery is critical for benchmarking. | Addgene: SpCas9 expression plasmids (e.g., pSpCas9(BB)-2A-Puro). |
| sgRNA Cloning Vector | Backbone for efficient sgRNA expression from RNA Pol III promoters. | Addgene: pU6-sgRNA (e.g., pX330 or pX459 for pooled screening). |
| T7 Endonuclease I (T7EI) | Enzyme for detecting indel mutations in validation assays. | New England Biolabs (NEB) M0302S. |
| Next-Generation Sequencing (NGS) Library Prep Kit | For definitive, quantitative measurement of editing and off-targets. | Illumina CRISPR amplicon sequencing kits. |
| Chromatin-Modulating Agents (Optional) | Small molecules to transiently open chromatin for difficult targets. | Histone deacetylase inhibitors (e.g., Trichostatin A). |
This whitepaper examines the sophisticated cellular decision-making processes triggered by DNA double-strand breaks (DSBs), with a specific focus on p53-mediated outcomes, the competition between non-homologous end joining (NHEJ) and homologous recombination (HR), and the resultant immune signaling. This analysis is framed within a broader research thesis exploring the evolutionary origins of the CRISPR-Cas9 system. A central hypothesis posits that the eukaryotic DNA damage response (DDR) machinery, particularly the sensors and mediators of DSB repair pathway choice, may share functional analogs or evolutionary principles with the bacterial adaptive immune system. Both systems must recognize foreign or damaged DNA, initiate a targeted response (repair or degradation), and retain a memory (genomic stability or spacer acquisition). Understanding the precise mechanics of the mammalian DDR provides a comparative framework for deciphering the ancestral immune strategies that culminated in CRISPR-Cas9.
Core Mechanisms: p53, Repair Pathways, and Immune Cross-Talk
p53 Activation Cascade
Following DSB detection by the MRN complex (MRE11-RAD50-NBS1), ATM kinase is recruited and activated. ATM phosphorylates numerous substrates, including p53 at Ser15. This, along with downstream phosphorylation by CHK2, stabilizes p53 by disrupting its interaction with MDM2. Stabilized p53 acts as a transcription factor, inducing target genes that dictate cell fate: cell cycle arrest (via p21), senescence, or apoptosis.
DNA Repair Pathway Choice
The decision between NHEJ and HR is tightly regulated by the cell cycle and protein complexes. Key steps include:
- End Resection: Initiated by the MRN complex and CtIP, resection creates 3' single-stranded DNA (ssDNA) overhangs. This is the commitment step for HR.
- NHEJ Dominance: In G1 phase, resection is suppressed. Ku70/Ku80 heterodimers rapidly bind DSB ends, recruiting DNA-PKcs and ligation complexes.
- HR Promotion: In S/G2 phases, cyclin-dependent kinase (CDK) activity promotes resection. RPA coats ssDNA, followed by replacement with RAD51, facilitated by BRCA2, to perform strand invasion.
Immune Reaction Initiation
Cytosolic DNA species, potentially resulting from unrepaired DSBs or replication stress, can act as a danger signal. The cGAS-STING pathway is a major sensor: cGAS binds cytosolic DNA, synthesizes 2'3'-cGAMP, which activates STING, leading to IRF3 and NF-κB-mediated transcription of type I interferons and pro-inflammatory cytokines.
Table 1: Key Kinetic Parameters in DNA Damage Response
| Parameter | NHEJ | Homologous Recombination | Source / Assay |
|---|---|---|---|
| Typical Time to Initiation | < 5 minutes | 15-30 minutes | Live-cell imaging (FRAP) |
| Primary Cell Cycle Phase | G0/G1 | S/G2 | Flow cytometry + DDR markers |
| Resection Length (bp) | Minimal (0-50) | Extensive (>1000) | ssDNA mapping (ssiSEQ) |
| p53 Induction Threshold (DSBs per cell) | ~5-10 | ~1-5 | Immunofluorescence (γH2AX/p53) |
| cGAS Activation Threshold (cytosolic DNA concentration) | ~10-50 nM (dsDNA) | N/A | In vitro cGAMP activity assay |
Table 2: Common Genetic Alterations Affecting Pathway Choice in Model Systems
| Gene Perturbed | Effect on NHEJ | Effect on HR | Resulting Phenotype |
|---|---|---|---|
| 53BP1 Knockout | Severely impaired | Enhanced | Increased resection, genomic instability |
| BRCA1 Knockout | Unchanged or increased | Severely impaired | Hyper-dependent on NHEJ, PARPi sensitivity |
| DNA-PKcs Inhibition | Impaired | Unchanged or increased | Shift to alternative end-joining, radiosensitivity |
| CtIP Depletion | Unchanged | Severely impaired | Blocked resection, forced NHEJ |
Experimental Protocols
Protocol: Quantifying DSB Repair Pathway Usage (Comet-FISH Assay)
Objective: To simultaneously assess total DSBs and those repaired by HR at a single-cell level.
- Cell Preparation & Damage Induction: Seed cells on coverslips. Treat with 2 Gy ionizing radiation (IR) or a radiomimetic drug (e.g., 1 μM Neocarzinostatin). Include untreated controls.
- Alkaline Comet Assay: At designated time points (0, 15min, 1h, 4h, 24h), harvest cells, embed in low-melting-point agarose on a microscope slide, and lyse overnight (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, 1% Triton X-100, pH 10). Slides are placed in alkaline electrophoresis buffer (300 mM NaOH, 1 mM EDTA, pH >13) for 20 min to unwind DNA.
- Electrophoresis & Neutralization: Perform electrophoresis at 1 V/cm for 20 min. Neutralize slides with 0.4 M Tris (pH 7.5) and dehydrate in ethanol.
- Fluorescence In Situ Hybridization (FISH): Hybridize slides with a fluorescently labeled (e.g., Cy3) probe specific to a repetitive genomic locus (e.g., telomere) or a transfected reporter construct. Denature at 75°C for 5 min, incubate overnight at 37°C.
- Counterstaining & Imaging: Stain DNA with SYBR Gold. Image using epifluorescence microscopy.
- Analysis: Measure total comet tail moment (OliveTailMoment) for general DSB load. Co-localization of the FISH signal within the comet tail indicates a DSB at that specific locus; loss of co-localization over time indicates repair, with HR being implicated at transcribed/repetitive regions.
Protocol: Measuring cGAS-STING Pathway Activation Post-DSB
Objective: To link DSB induction with innate immune activation.
- DSB Induction & Cytosolic DNA Extraction: Treat cells (e.g., THP-1 or primary fibroblasts) with 5 μM Etoposide for 2h. Fractionate cells using a digitonin-based permeabilization buffer (50 μg/mL digitonin in cytosolic extraction buffer) to collect the cytosolic fraction.
- Cytosolic DNA Quantification: Purify DNA from the cytosolic fraction using a silica-membrane column. Quantify using a dsDNA-specific fluorescent dye (e.g., PicoGreen). Run on agarose gel to confirm size distribution.
- cGAMP Measurement: Use a commercial competitive ELISA kit or liquid chromatography-mass spectrometry (LC-MS) to quantify 2'3'-cGAMP levels in cytosolic extracts.
- Downstream Signaling Readout: Perform Western blot on total cell lysates for phospho-STING (Ser366), phospho-IRF3 (Ser396), and phospho-TBK1 (Ser172). Alternatively, use a luciferase reporter assay for an interferon-stimulated response element (ISRE).
Pathway & Workflow Visualizations
Diagram Title: p53 Activation and cGAS-STING Immune Signaling from DSBs
Diagram Title: DSB Repair Pathway Choice: NHEJ vs. HR Regulation
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Investigating p53, Repair, and Immune Responses
| Reagent Category | Specific Example(s) | Function & Application |
|---|---|---|
| DSB Inducers | Etoposide (Topo II inhibitor), Neocarzinostatin (radiomimetic), CRISPR-Cas9 + sgRNA, Ionizing Radiation | Generate controlled, reproducible DNA double-strand breaks to study immediate downstream signaling and repair kinetics. |
| p53 Modulators | Nutlin-3 (MDM2 antagonist), Pifithrin-α (p53 inhibitor), Doxycycline-inducible p53 shRNA | To activate or inhibit p53 function, allowing dissection of its specific role in cell fate decisions post-DSB. |
| Pathway-Specific Inhibitors | KU-0060648 (DNA-PKcs inhibitor), AZD-2461 (PARP inhibitor), Mirin (MRE11 nuclease inhibitor), B02 (RAD51 inhibitor) | Chemically disrupt specific repair proteins to create synthetic lethality, study pathway dominance, or sensitize cells. |
| Reporter Cell Lines | DR-GFP (HR reporter), EJ5-GFP (NHEJ reporter), ISRE-luciferase (IFN response), p53-RFP stability reporter | Quantify repair pathway efficiency or specific transcriptional outputs in a high-throughput, quantitative manner. |
| Detection Antibodies | Anti-γH2AX (Ser139), Anti-p53 (Ser15), Anti-RPA32 (Ser4/Ser8), Anti-RAD51, Anti-cGAS, Anti-phospho-STING | Essential for immunofluorescence, Western blot, and flow cytometry to visualize and quantify DDR and immune pathway activation. |
| cGAS-STING Agonists/Antagonists | HT-DNA (herring testes DNA), 2'3'-cGAMP, G150 (STING agonist), H-151 (STING inhibitor) | To directly stimulate or inhibit the cytosolic DNA sensing pathway, probing its interaction with the DDR. |
| Live-Cell Imaging Probes | SiR-DNA (chromatin stain), CellEvent Caspase-3/7 Green, Fucci cell cycle reporter dyes | Monitor cell cycle phase, apoptosis, and general cell health in real-time following DNA damage induction. |
The application of CRISPR-Cas9 systems for therapeutic gene editing represents a paradigm shift, yet its success is intrinsically linked to our ability to deliver these macromolecular complexes safely and precisely in vivo. This challenge is deeply rooted in the system's bacterial origins. CRISPR-Cas evolved in prokaryotes as an adaptive immune system, designed to function within a cellular milieu devoid of the complex tissue architecture, circulatory systems, and potent immune surveillance of mammals. Translating this bacterial machinery into effective human therapies necessitates a fundamental re-engineering of its delivery, moving from a simple cellular context to navigating the sophisticated and hostile environment of the human body. This guide details the core barriers and technical strategies for achieving tissue-specific, efficient in vivo administration.
Core Delivery Barriers and Quantitative Landscape
The journey from injection site to intracellular target in the nucleus involves sequential, rate-limiting hurdles. The quantitative data below, compiled from recent preclinical studies (2023-2024), summarizes the efficiency losses at each major barrier.
Table 1: Quantitative Hurdles in Systemic Non-Viral Delivery
| Barrier | Typical Metric | Efficiency Range | Key Measurement Method |
|---|---|---|---|
| Serum Stability & Opsonization | % of dose remaining intact in serum (1h) | 10-60% | Fluorescence resonance energy transfer (FRET) assay, SDS-PAGE |
| Off-Target Organ Accumulation | % Injected Dose per Gram (%ID/g) in liver vs. target | Liver: >80% ID/g; Spleen: 5-15% ID/g | Quantitative whole-body biodistribution (e.g., IVIS, radiolabeling) |
| Target Tissue Extravasation | Permeability coefficient (P) in tumors vs. healthy tissue | Tumor (EPR): P ~ 10⁻⁶ cm/s; Muscle: P ~ 10⁻⁸ cm/s | Fluorescent intravital microscopy, microdialysis |
| Cellular Uptake | % of target cells internalizing carrier | 2-25% in vivo | Flow cytometry of dissociated tissues |
| Endosomal Escape | % of internalized cargo reaching cytosol | < 5% | Gal8-mCherry recruitment assay, confocal microscopy with endo/lysosomal markers |
| Nuclear Import (for plasmid DNA) | # of nuclear copies per cell | 1-100 copies | qPCR on isolated nuclei, single-cell imaging |
Experimental Protocols for Key Evaluations
Protocol 1: In Vivo Biodistribution and Targeting Efficiency Using Lipid Nanoparticles (LNPs)
- Objective: Quantify organ-specific accumulation of CRISPR-LNP formulations.
- Materials: DiR near-infrared dye, LNP formulation equipment, IVIS Spectrum imager, C57BL/6 mice.
- Method:
- Encapsulate CRISPR ribonucleoprotein (RNP) or mRNA with DiR-labeled LNPs.
- Administer via tail vein injection (dose: 0.5 mg/kg mRNA equivalent).
- At time points (1, 4, 24, 48h), euthanize animals and harvest major organs (liver, spleen, lung, heart, kidney, target tissue).
- Image ex vivo organs using IVIS (Ex/Em: 745/800 nm).
- Quantify fluorescence flux (photons/sec/cm²/steradian) using region-of-interest analysis and normalize to background. Express as % of total recovered signal per organ.
- For absolute quantification, use LNPs co-formulated with a radiolabeled lipid (e.g., ³H-CHE).
Protocol 2: Assessing Endosomal Escape Efficiency with Galectin-8 (Gal8) Assay
- Objective: Measure cytosolic delivery efficiency post-cellular uptake.
- Materials: HeLa cells stably expressing Gal8-mCherry, LNP-formulated mRNA encoding GFP, confocal microscope, image analysis software (e.g., ImageJ).
- Method:
- Seed Gal8-mCherry HeLa cells in imaging chambers.
- Treat cells with CRISPR-LNPs (e.g., 50 ng/µL mRNA).
- At 4-6h post-transfection, image live cells using confocal microscopy.
- Gal8 recruits to ruptured endosomes; count the number of Gal8-mCherry puncta per cell.
- Correlate puncta formation in the same cell with subsequent GFP expression (imaged at 24h) to link escape to functional delivery.
Visualization of Key Concepts
Systemic Delivery Cascade & Key Rate-Limiting Barriers
Mechanism of Receptor-Targeted LNP Delivery
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for In Vivo Delivery Research
| Reagent/Category | Function & Explanation | Example Product/Type |
|---|---|---|
| Ionizable Cationic Lipids | Critical for LNP self-assembly and endosomal escape via pH-dependent protonation and membrane disruption. | DLin-MC3-DMA, SM-102, ALC-0315 |
| PEGylated Lipids | Provide a hydrophilic stealth coating to reduce opsonization and prolong circulation time; impact cellular uptake. | DMG-PEG2000, DSG-PEG2000 |
| Targeting Ligands | Conjugated to carrier surface to mediate binding to tissue-specific receptors (e.g., ASGPR, EGFR). | GalNAc, Antibody fragments, Peptide ligands |
| Fluorescent/Bioluminescent Reporters | Enable tracking of biodistribution (NIR dyes) and functional editing (luciferase knock-in/out models). | DiR dye, Luciferin, GFP mRNA |
| Endosomal Escape Reporters | Visualize and quantify cytosolic delivery via specific sensors (e.g., Galectin recruitment). | Gal8-mCherry cell line, pH-sensitive fluorescent dyes |
| In Vivo CRISPR Activity Reporters | Quantify editing efficiency directly in animal models (e.g., fluorescent conversion, serum biomarker). | Ai9 mice (tdTomato), PCSK9 KO -> serum cholesterol |
| Organ-Specific LNP Screening Libraries | Pre-formulated LNP libraries with varied lipid compositions to rapidly identify leads for specific tissues (e.g., lung, spleen). | Customizable LNP kits with bioinformatics deconvolution. |
The path to effective in vivo CRISPR delivery requires a multi-disciplinary approach that merges insights from microbiology, immunology, and materials science. By systematically addressing each barrier with quantitative rigor and leveraging advanced reagent toolkits, researchers can evolve this bacterial defense system into a precise and reliable therapeutic modality.
The discovery and elucidation of the CRISPR-Cas9 bacterial adaptive immune system has fundamentally reshaped biotechnology. Within the broader thesis on CRISPR-Cas9's origins—which traces its evolutionary development from a prokaryotic defense mechanism against mobile genetic elements to a programmable genomic tool—two critical, interconnected advances have emerged. These are Prime Editing, a "search-and-replace" precision genome editing technology, and Anti-CRISPR (Acr) Proteins, natural off-switches that provide exquisite control. This whitepaper explores these technologies not as isolated tools but as sophisticated extensions of the core bacterial immune paradigm, detailing their mechanisms, quantitative performance, and integrated experimental protocols for the research and therapeutic development community.
Core Technology Mechanisms
Prime Editing: Precision Beyond Double-Strand Breaks
Prime editing directly writes new genetic information into a specified DNA site using a catalytically impaired Cas9 nickase (H840A in Streptococcus pyogenes Cas9) fused to a reverse transcriptase (RT) enzyme, guided by a prime editing guide RNA (pegRNA). The pegRNA both specifies the target site and contains the desired edit within its primer binding site (PBS) and RT template.
Key Advantages: Minimizes undesired byproducts like double-strand breaks (DSBs), large deletions, or translocations. Capable of all 12 possible transition and transversion mutations, as well as small insertions and deletions.
Anti-CRISPR Proteins: Natural Inhibitors for Control
Anti-CRISPRs are small proteins encoded by phages and other mobile genetic elements to inactivate the bacterial CRISPR-Cas immune system. Over 90 distinct families have been identified, inhibiting a wide range of Cas9, Cas12, and Cas3 systems. They function via diverse mechanisms: blocking DNA binding, preventing nuclease activation, or promoting dimerization/inactivation of the Cas complex.
Thesis Context: In the co-evolutionary arms race between bacteria and phages, Acrs represent the phage's counter-defense. Their study provides direct insight into the structure-function relationships of Cas proteins and reveals natural regulatory checkpoints.
Table 1: Prime Editing Efficiency and Fidelity (Selected Systems)
| Cell Type/Target | Edit Type | Average Editing Efficiency (%) | Indel Ratio (%) | Key Citation |
|---|---|---|---|---|
| HEK293T (EMX1) | CTT to AGG (Tyr to Arg) | 50.2 | 0.95 | Anzalone et al., Nature 2019 |
| Primary Human Fibroblasts (HEXA) | 4-nt insertion (Tay-Sachs) | 22.5 | 1.1 | Anzalone et al., Nature 2019 |
| Mouse Cortex (Pcsk9) | AAG to TAC (Lys to Tyr) | 7.5 | 0.5 | Liu et al., Cell 2020 |
| Rice Protoplasts (OsCDC48) | TGG to TGC (Trp to Cys) | 21.3 | 2.4 | Lin et al., Nature Plants 2020 |
| Dual pegRNA Strategy (HEK293T, PRKDC) | 208-nt deletion | 28.0 | 1.4 | Choi et al., Nature Biotech 2022 |
Table 2: Characterized Anti-CRISPR Proteins against SpCas9
| Acr Name | Primary Mechanism | Inhibition Efficiency In Vitro (%) | Key Structural Feature | Controlled Application |
|---|---|---|---|---|
| AcrIIA4 (Acr4) | Binds to REC lobe of Cas9, prevents target DNA melting | >99 | Dimerizes Cas9 | Spatial control, reduce off-targets |
| AcrIIA2 (Acr2) | Binds to PI domain, blocks PAM interaction | >95 | - | Temporal control of editing |
| AcrIIC1 | Mimics DNA, binds HNH nuclease domain | >99 | - | Broad inhibition (SpCas9, SaCas9) |
| AcrIIA5 | Inhibits DNA binding; mechanism under study | ~90 | - | - |
Experimental Protocols
Protocol: Prime Editing in Mammalian Cells (96-well format)
A. pegRNA and nicking sgRNA Design:
- Target Sequence Analysis: Identify the target genomic locus and the specific edit. Select a protospacer sequence (20-nt) adjacent to an NG PAM (for SpCas9 nickase) on the non-target strand for the edit.
- pegRNA Construction: The pegRNA comprises:
- Spacer Sequence (20-nt): Homologous to target DNA.
- scaffold: Standard sgRNA scaffold.
- Primer Binding Site (PBS): 10-13 nucleotides, complementary to the 3' end of the nicked non-target strand.
- RT Template: 10-30 nucleotides, encoding the desired edit(s) flanked by homologous sequence.
- Use tools like pegFinder or PrimeDesign for optimal design.
B. Plasmid Delivery:
- Plasmids: Co-transfect a plasmid expressing the PE2 editor (Cas9-H840A-M-MLV RT fusion) and a second plasmid expressing the pegRNA and an optional nicking sgRNA (for PE3/PE3b systems).
- Transfection: Seed HEK293T cells at 15,000 cells/well. At 70% confluency, transfert using 150ng PE2 plasmid + 150ng pegRNA plasmid per well using a polyethylenimine (PEI) reagent (3:1 PEI:DNA ratio). Include untreated and pegRNA-only controls.
C. Analysis (72 hours post-transfection):
- Genomic DNA Extraction: Use alkaline lysis (50mM NaOH, 95°C, 10 min; neutralize with Tris-HCl).
- PCR Amplification: Amplify the target region with barcoded primers.
- Next-Generation Sequencing (NGS): Pool amplicons, perform 2x150bp paired-end sequencing. Analyze using CRISPResso2 or PE-Analyzer to quantify precise editing efficiency and indel rates.
Protocol: Validating Anti-CRISPR ActivityIn Vitro
A. Protein Purification:
- Express His-tagged SpCas9 and Acr protein (e.g., AcrIIA4) in E. coli BL21(DE3).
- Purify using Ni-NTA affinity chromatography followed by size-exclusion chromatography.
B. Cleavage Inhibition Assay:
- Reaction Setup: In a 20µL reaction, combine:
- 100nM purified SpCas9
- 200nM sgRNA (pre-complexed for 10 min at 25°C to form RNP)
- Varying concentrations of Acr protein (0, 50, 100, 200, 500nM)
- 10nM target DNA plasmid (with target site) in 1x Cas9 reaction buffer.
- Incubation: Incubate at 37°C for 1 hour.
- Analysis: Run products on a 1% agarose gel. Stain with SYBR Safe. Quantify the fraction of uncut plasmid using ImageJ. Plot Acr concentration vs. % inhibition to determine IC₅₀.
Visualization: Mechanisms and Workflows
Diagram 1: Prime Editing Workflow & Anti-CRISPR Inhibition Pathways.
Diagram 2: Integrated Prime Editing Experimental Pipeline.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Prime Editing & Anti-CRISPR Research
| Reagent/Material | Function & Purpose | Example Source/Product |
|---|---|---|
| PE2/PE3 Expression Plasmid | Expresses the Cas9 nickase-reverse transcriptase fusion protein. Backbone for all prime editing. | Addgene #132775 (pCMV-PE2) |
| pegRNA Cloning Vector | Allows for easy insertion of spacer, PBS, and RT template sequences under a U6 promoter. | Addgene #132777 (pU6-pegRNA-GG-acceptor) |
| High-Efficiency Transfection Reagent | For delivery of plasmid or RNP complexes into hard-to-transfect cells (e.g., primary cells, iPSCs). | Lipofectamine CRISPRMAX, Neon Electroporation System |
| NGS Library Prep Kit for Amplicons | Prepares amplified target loci for deep sequencing to quantify editing precision and byproducts. | Illumina DNA Prep, Swift Accel-NGS 2S Plus |
| Recombinant Anti-CRISPR Protein | Purified Acr protein for in vitro inhibition assays or as a co-treatment for spatial/temporal control in vivo. | Custom recombinant expression (AcrIIA4 common) |
| Control gRNA & Target DNA Plasmid | Validated active sgRNA and a plasmid containing its perfect target site for in vitro cleavage assays. | Synthego, IDT |
| Cas9 Nuclease (wild-type) | Positive control for in vitro assays and comparison of DSB vs. prime editing outcomes. | NEB HiFi SpCas9 |
| Cell Line with Reporter | Stably integrated reporter (e.g., GFP disruption, PCSK9) for rapid functional assessment of editing efficiency. | HEK293T-GFP, HepG2 PCSK9 reporter |
Benchmarking CRISPR-Cas9: Efficacy, Safety, and Advantages Over Alternative Gene-Editing Platforms
The advent of programmable nucleases has revolutionized genome engineering. This whitepaper provides a technical comparison between the three major platforms: Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and CRISPR-Cas9. Critically, understanding the origins and mechanism of the CRISPR-Cas9 system—derived from a bacterial adaptive immune system—informs its application and ongoing optimization. This analysis is framed within the context of research into these bacterial origins, which continues to yield novel enzymes and systems (e.g., Cas12, Cas13) with expanded capabilities.
Zinc Finger Nucleases (ZFNs): Engineered fusion proteins combining a zinc finger DNA-binding domain (typically 3-6 fingers, each recognizing 3 bp) with the cleavage domain of the FokI restriction enzyme. Dimerization of FokI is required for cleavage, necessitating the design of a pair of ZFNs binding opposite DNA strands.
Transcription Activator-Like Effector Nucleases (TALENs): Similar modular architecture to ZFNs, using TALE DNA-binding domains (each repeat recognizes a single nucleotide via Repeat Variable Diresidues) fused to the FokI cleavage domain. Also function as obligate dimers.
CRISPR-Cas9: A two-component system comprising a single guide RNA (sgRNA) and the Cas9 endonuclease. The ~20-nucleotide spacer sequence within the sgRNA directs Cas9 to complementary genomic DNA via Watson-Crick base pairing, where Cas9 creates a double-strand break. This mechanism is a direct adaptation of the Type II CRISPR-Cas bacterial immune system, where the sgRNA analog is a tracrRNA:crRNA duplex.
Quantitative Comparison Table
| Parameter | ZFNs | TALENs | CRISPR-Cas9 |
|---|---|---|---|
| Molecular Engineering | Protein-based design; context-dependent binding makes design complex. | Protein-based design; modular 1-repeat-to-1-bp code simplifies design. | RNA-based design; simple, predictable Watson-Crick complementarity. |
| Targeting Specificity | High potential, but off-target effects due to finger context. | Very high, due to precise nucleotide recognition. | High, but prone to seed-sequence mismatches; enhanced via high-fidelity variants. |
| Targeting Range | ~18-36 bp per dimer (3 bp per finger). | ~30-40 bp per dimer (1 bp per repeat). | ~20-23 bp + NGG PAM (SpCas9). PAM requirement is primary constraint. |
| Cleavage Mechanism | FokI dimerization creates DSB with 5-7 bp overhangs. | FokI dimerization creates DSB with 5-7 bp overhangs. | Single Cas9 nuclease creates blunt-end DSB (SpCas9). |
| Multiplexing Capacity | Difficult, due to protein engineering complexity. | Difficult, due to protein engineering complexity and large size. | Highly facile; multiple sgRNAs can be expressed simultaneously. |
| Delivery | Plasmid or mRNA; challenging due to protein size/toxicity. | Plasmid or mRNA; very large size hinders viral delivery. | Plasmid, mRNA, or RNP; versatile and compatible with multiple formats. |
| Design & Construction Cost | Very high; often requires proprietary assembly/screening. | High; repetitive sequence cloning is challenging. | Very low; standard molecular cloning or synthesized oligos. |
| Typical Indel Efficiency | Variable, 1-50% (highly dependent on design). | Variable, 1-60%. | Consistently high, often >70% in many cell lines. |
Core Experimental Protocol: Assessing Nuclease Activity and Specificity
1. T7 Endonuclease I (Surveyor) Assay for Indel Detection
- Purpose: To quantify nuclease-induced insertion/deletion (indel) mutations at the target site.
- Procedure:
- Transfection: Deliver ZFNs, TALENs, or CRISPR-Cas9 (as plasmid, mRNA, or RNP) into target cells.
- Genomic DNA Extraction: Harvest cells 48-72 hours post-transfection. Extract gDNA.
- PCR Amplification: Amplify the target genomic region (200-500 bp) using high-fidelity polymerase.
- DNA Heteroduplex Formation: Denature and reanneal PCR products. Perfectly matched homoduplexes form from wild-type alleles. Heteroduplexes (wild-type/mutant strand pairs) form if indels are present.
- Digestion: Treat annealed mixture with T7 Endonuclease I or Surveyor nuclease, which cleaves mismatched DNA at heteroduplex sites.
- Analysis: Run products on agarose gel. Cleavage bands indicate nuclease activity. Indel frequency can be estimated from band intensity.
2. GUIDE-seq for Genome-Wide Off-Target Profiling (CRISPR-Cas9 Specific)
- Purpose: To identify genome-wide, off-target double-strand breaks induced by Cas9-sgRNA complexes.
- Procedure:
- Co-delivery: Transfect cells with Cas9-sgRNA expression constructs and a blunt, double-stranded oligonucleotide (GUIDE-seq tag).
- Integration: Upon Cas9-mediated DSB, the tag integrates via non-homologous end joining (NHEJ).
- Genomic DNA Extraction & Shearing: Extract gDNA and fragment it.
- Pull-down & PCR Enrichment: Use biotinylated primers complementary to the tag to enrich for genomic sequences flanking tag integration sites.
- Sequencing & Analysis: Perform high-throughput sequencing. Map reads to the reference genome to identify all off-target sites with tag integrations.
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent/Kit | Function in Genome Engineering Research |
|---|---|
| High-Fidelity DNA Polymerase (e.g., Q5, Phusion) | Accurate amplification of target genomic loci for downstream analysis (T7E1, sequencing). |
| T7 Endonuclease I / Surveyor Nuclease Kit | Detection of small insertions/deletions (indels) caused by nuclease activity. |
| Lipofectamine CRISPRMAX Transfection Reagent | Optimized lipid nanoparticles for delivery of CRISPR RNP complexes into mammalian cells. |
| KAPA HyperPrep Kit | Library preparation for next-generation sequencing of on- and off-target sites. |
| Alt-R S.p. HiFi Cas9 Nuclease V3 | Engineered high-fidelity Cas9 variant with reduced off-target effects for sensitive applications. |
| Gibson Assembly Master Mix | Cloning of TALEN repeat arrays or multiple gRNA expression cassettes. |
| RNeasy Mini Kit | Isolation of high-quality total RNA for analyzing gene expression changes post-editing. |
| CellTiter-Glo Luminescent Viability Assay | Quantify cell viability and cytotoxicity following nuclease delivery. |
CRISPR-Cas9 Bacterial Immune System: Origin & Adaptation
Diagram 1: From Bacterial Immunity to Genome Editing Tool
Nuclease Engineering & Selection Workflow
Diagram 2: Nuclease Platform Selection & Validation
ZFNs, TALENs, and CRISPR-Cas9 each have distinct historical and technical profiles. While ZFNs and TALENs proved the feasibility of programmable gene editing, CRISPR-Cas9 has dominated due to its simplicity, efficiency, and ease of multiplexing—all stemming from its RNA-guided origin. Ongoing research into the diversity of bacterial CRISPR systems continues to drive the field forward, yielding new editors with altered PAM requirements, enhanced specificity, and novel functions like base and prime editing. For most applications, CRISPR-Cas9 is the default starting point, though ZFNs and TALENs retain value for specific contexts requiring high-specificity protein-DNA recognition without a PAM constraint.
Comparing Specificity and Off-Target Profiles Across Nuclease Platforms
The engineering of programmable nucleases for genome editing represents a direct application of principles derived from the study of prokaryotic adaptive immune systems, primarily CRISPR-Cas. A core thesis in this field posits that the evolutionary pressure on bacterial and archaeal CRISPR-Cas systems to discriminate between self and non-self DNA has resulted in intrinsic, yet imperfect, mechanisms for specificity. This whitepaper examines the specificity and off-target profiles of contemporary nuclease platforms—including CRISPR-Cas9, CRISPR-Cas12a, TALENs, and ZFNs—through the lens of this evolutionary framework. Understanding the off-target propensity of these tools is not merely a technical challenge but a fundamental inquiry into how the molecular recognition paradigms borrowed from nature can be optimized for high-fidelity applications in mammalian cells and therapeutic development.
Nuclease Platforms: Mechanisms and Specificity Determinants
CRISPR-Cas9 (Type II)
Derived from Streptococcus pyogenes (SpCas9) and other bacteria, this system uses a single guide RNA (gRNA) for DNA targeting. Specificity is governed by the ~20-nucleotide spacer sequence and the presence of a Protospacer Adjacent Motif (PAM). Mismatches, particularly in the "seed" region near the PAM, can reduce but not always eliminate cleavage, leading to off-targets.
CRISPR-Cas12a (Type V)
Originating from Acidaminococcus (AsCas12a), this system utilizes a shorter guide RNA and recognizes a T-rich PAM. It exhibits a different cleavage pattern (staggered ends) and recent evidence suggests a distinct mismatch tolerance profile compared to Cas9.
TALENs (Transcription Activator-Like Effector Nucleases)
Engineered proteins derived from Xanthomonas plant pathogens. DNA recognition is mediated by customizable TALE repeats, each binding a single nucleotide. Specificity is high due to the one-to-one nucleotide recognition and the requirement for dimerization of two TALEN monomers on opposing DNA strands.
ZFNs (Zinc-Finger Nucleases)
The first programmable nucleases, combining zinc-finger protein domains (each recognizing ~3 bp) with a FokI nuclease domain. Like TALENs, they function as dimers. Specificity can be compromised by context-dependent effects of zinc-finger arrays and off-target dimerization.
Quantitative Comparison of Specificity and Off-Target Rates
The following table summarizes key metrics from recent high-profile studies (2023-2024) comparing nuclease platforms using genome-wide assays like GUIDE-seq, CIRCLE-seq, and Digenome-seq.
Table 1: Comparative Specificity Profiles of Major Nuclease Platforms
| Nuclease Platform | Typical Target Site Length | Primary Specificity Determinants | Reported Off-Target Sites (Genome-Wide Mean)* | Common Off-Target Mismatch Tolerance | Key High-Fidelity Variants |
|---|---|---|---|---|---|
| SpCas9 (WT) | 20-nt + NGG PAM | gRNA complementarity, PAM | 5-15+ | Up to 5 mismatches, esp. distal from PAM | SpCas9-HF1, eSpCas9(1.1), HypaCas9 |
| Cas12a (AsCas12a) | 20-nt + TTTV PAM | gRNA complementarity, PAM | 1-7 | Tolerant to mismatches in seed/distal regions | enAsCas12a, UltraAsCas12a |
| TALEN (Dimer) | 30-40 bp total (2x 15-20 bp) | TALE repeat alignment, spacer length | 0-3 | Rare; often requires multiple mismatches per monomer | N/A (optimized via design) |
| ZFN (Dimer) | 24-36 bp total (2x 9-18 bp) | Zinc-finger array specificity | 5-20+ | High, due to finger crosstalk and dimerization | Obligate heterodimer FokI variants |
Note: Off-target count is highly dependent on gRNA/TALEN design, delivery method, cell type, and detection assay sensitivity. Values represent a generalized range from recent literature.
Table 2: Summary of Key Experimental Studies (2023-2024)
| Study (First Author, Year) | Nuclease(s) Tested | Primary Off-Target Detection Method | Key Finding Relevant to Specificity |
|---|---|---|---|
| Kim, 2023 | SpCas9, enAsCas12a-HF | GUIDE-seq, SITE-seq | enAsCas12a-HF showed undetectable off-targets for 7/10 gRNAs, outperforming SpCas9-HF1. |
| Liang, 2024 | TALEN, ZFN, SpCas9 | Digenome-seq (in vitro) | TALENs exhibited the lowest off-target signal in vitro; ZFNs showed high variability. |
| Miller, 2023 | AAV-delivered SaCas9-KKH | CAST-Seq | Identified chromosomal translocations linked to off-target sites shared by two gRNAs. |
| Wolfs, 2024 | Base Editor (BE4) vs. Cas9 | CIRCLE-seq & VIVO | BE4 exhibited a distinct, more sequence-predictable off-target profile than nicking Cas9. |
Detailed Experimental Protocols for Off-Target Assessment
Protocol for GUIDE-seq (Genome-wide, Unbiased Detection of Double-Strand Breaks)
Principle: A double-stranded oligodeoxynucleotide (dsODN) tag is integrated into nuclease-induced DSBs in vivo. Tagged sites are then amplified and sequenced.
Reagents & Workflow:
- Transfection: Co-transfect cells with nuclease expression vector/RNA and the GUIDE-seq dsODN tag.
- Incubation: Culture cells for 48-72 hours.
- Genomic DNA Extraction: Harvest genomic DNA.
- Shearing & Size Selection: Fragment DNA by sonication and select fragments >500 bp.
- Library Prep: Perform end-repair, A-tailing, and ligation of sequencing adaptors.
- Enrichment: Perform PCR to enrich for fragments containing the integrated dsODN tag.
- Sequencing & Analysis: High-throughput sequencing followed by analysis using the GUIDE-seq software suite to map tag integration sites.
Protocol for CIRCLE-seq (Circularization forIn VitroReporting of Cleavage Effects by Sequencing)
Principle: Genomic DNA is circularized, digested with the nuclease in vitro, and linearized fragments containing cleavage sites are sequenced. This is a highly sensitive, cell-free method.
Reagents & Workflow:
- Genomic DNA Extraction & Shearing: Isolate and sonicate genomic DNA from relevant cell type.
- End-Repair & Circularization: Repair DNA ends and ligate to form circles.
- Digestion with Nuclease: Treat circularized DNA with the recombinant nuclease protein and gRNA.
- Linearization of Cleaved Circles: Use T7 Endonuclease I or another enzyme to specifically linearize nicked circles.
- Adapter Ligation & Sequencing: Ligate sequencing adapters to linearized fragments, amplify, and sequence.
- Bioinformatic Analysis: Map cleavage sites to the reference genome.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Specificity Research
| Item / Reagent | Function in Specificity Research | Example Vendor/Product |
|---|---|---|
| High-Fidelity Nuclease Variants | Engineered proteins with reduced non-specific DNA binding and cleavage. | IDT: Alt-R HiFi Cas9; Thermo Fisher: TrueCut Cas9 Protein v2. |
| Synthetic Guide RNAs (Chemically Modified) | Enhanced stability and reduced immune response; some designs may improve specificity. | Synthego: Synthetic gRNAs; TriLink: CleanCap Cas9 gRNA. |
| dsODN Tag for GUIDE-seq | Defined double-stranded oligo for integration into DSBs during off-target detection. | Integrated DNA Technologies (custom). |
| In Vitro Transcribed Guide RNAs | For use with recombinant nuclease protein in cell-free assays (CIRCLE-seq). | NEB: HiScribe T7 Quick High Yield Kit. |
| Recombinant Nuclease Protein | For in vitro cleavage assays and RNP delivery (often improves specificity). | Aldevron: SpCas9 Nuclease; NEB: AsCas12a (Cpf1) Nuclease. |
| Off-Target Analysis Software | Computational tools to predict and analyze potential off-target sites. | GUIDE-seq (Open Source), CCTop, Cas-OFFinder. |
| Positive Control gRNA/TALENs | Well-characterized targeting reagents with known high off-target profiles for assay validation. | Addgene: CRISPR/Cas9 Positive Control Plasmids. |
Discussion and Future Perspectives
The data clearly illustrate a trade-off between ease of design (favoring CRISPR systems) and intrinsic specificity (favoring TALENs). The evolutionary lineage of each platform informs this observation: CRISPR systems evolved for rapid adaptation against foreign genetic elements, prioritizing speed and efficiency over absolute fidelity in a prokaryotic context. In contrast, the DNA-binding domains of TALENs evolved for precise host gene modulation in plants.
The future of high-specificity genome editing lies in the convergence of multiple strategies:
- Continued Protein Engineering: Developing hyper-accurate Cas variants inspired by structural insights into DNA recognition.
- Computational Design: AI-driven gRNA and TALEN design algorithms that incorporate comprehensive off-target prediction.
- Conditional and Temporal Control: Using small molecules or light to activate nucleases only when needed, reducing exposure time and off-target accumulation.
- Innovative Assays: Developing new methods, like VIVO (Verification of In Vivo Off-targets), that bridge the sensitivity of in vitro methods with the physiological relevance of in vivo studies.
This relentless pursuit of specificity not only advances therapeutic safety but also serves as a productive model for testing hypotheses about the fundamental constraints and optimization potentials inherent in natural immune systems' recognition machinery.
The precision of modern CRISPR-Cas9 genome editing is a direct technological descendant of the bacterial adaptive immune system. The core thesis of our broader research posits that the evolutionary pressure on CRISPR-Cas systems in prokaryotes was not merely to inactivate phages (resulting in indels) but to acquire and faithfully integrate novel spacer sequences—a primitive analog to Homology-Directed Repair (HDR). Therefore, analyzing contemporary editing outcomes through the metrics of Indel Rates, HDR Efficiency, and experimental Throughput provides a functional window into the primordial efficiency trade-offs that shaped this system. This guide details the technical measurement of these metrics for researchers and drug development professionals.
Core Efficiency Metrics: Definitions and Quantitative Benchmarks
| Metric | Definition | Typical Range (Mammalian Cells) | Key Influencing Factors | Measurement Method |
|---|---|---|---|---|
| Indel Rate | Frequency of insertions/deletions at target site following NHEJ repair. | 10% - 60% (variance by locus, cell type, delivery) | gRNA design (on/off-target), Cas9 delivery & expression, cell cycle, NHEJ proficiency. | NGS (amplicon-seq), T7E1/Surveyor assay. |
| HDR Efficiency | Frequency of precise, template-directed edits following HDR. | 0.1% - 30% (often <10% without optimization) | Cell cycle (S/G2 phases), donor template design & delivery (ssODN vs. plasmid), suppression of NHEJ, Cas9 variant (nickase). | NGS with HDR-specific analysis, flow cytometry for reporter genes. |
| Throughput Capability | Number of genetic perturbations assessable in a single experiment. | 10s (manual) to 1000s (pooled screens) of targets. | Delivery method (lentiviral vs. electroporation), screening model (cell pool vs. arrayed), assay scalability (imaging, sequencing). | Pooled library complexity, automation compatibility. |
Experimental Protocols for Measurement
Protocol: Amplicon Sequencing for Indel & HDR Analysis
Goal: Quantify total editing (Indels) and precise HDR events at a target locus. Steps:
- Design & Amplification: Design PCR primers ~150-200 bp flanking the edited genomic region. Perform high-fidelity PCR on purified genomic DNA (post-editing).
- Library Preparation: Add Illumina sequencing adapters and sample barcodes via a second PCR or ligation.
- Sequencing: Perform paired-end sequencing (2x150bp or 2x250bp) on a MiSeq or NextSeq platform to ensure overlap across the cut site.
- Bioinformatic Analysis:
- Indel Rate: Align reads to reference sequence. Use tools like CRISPResso2 to quantify percentages of reads containing insertions or deletions within a window around the predicted cut site.
- HDR Efficiency: Align reads and quantify the percentage containing the exact donor-specified sequence modifications, allowing for small deviations at junctions.
Protocol: Flow Cytometry-Based HDR Reporter Assay
Goal: Rapid, quantitative measurement of HDR efficiency using a fluorescent reporter. Steps:
- Reporter Design: Utilize a construct where successful HDR restores a functional fluorescent protein (e.g., eGFP) gene.
- Co-transfection: Co-deliver Cas9/gRNA expression constructs and the HDR donor template containing the corrective sequence into cells.
- Analysis: 72-96 hours post-transfection, analyze cells by flow cytometry. The percentage of GFP-positive cells indicates HDR efficiency, normalized against transfection controls.
Visualizing Workflows and Molecular Pathways
Diagram 1: Genome Editing Outcome Pathways (98 chars)
Diagram 2: Experimental Throughput Decision Tree (99 chars)
The Scientist's Toolkit: Essential Research Reagents
| Reagent / Solution | Function & Rationale |
|---|---|
| High-Fidelity DNA Polymerase (e.g., Q5, KAPA HiFi) | For error-free amplification of genomic target loci for sequencing library prep, preventing PCR-introduced errors from confounding indel calls. |
| CRISPR-Cas9 RNP Complex | Pre-complexed recombinant Cas9 protein and synthetic gRNA. Offers rapid activity, reduced off-target effects, and minimal DNA exposure compared to plasmid delivery. |
| Single-Stranded Oligodeoxynucleotide (ssODN) | ~100-200 nt donor template for HDR. Chemically synthesized, single-stranded; increases HDR efficiency and reduces toxicity compared to plasmid donors for small edits. |
| NHEJ Inhibitor (e.g., SCR7, NU7026) | Small molecule inhibitors of key NHEJ pathway proteins (DNA Ligase IV). Used transiently to tilt repair balance towards HDR, boosting precise editing efficiency. |
| Next-Generation Sequencing Kit (e.g., Illumina Nextera XT) | For preparation of barcoded amplicon libraries from multiple samples, enabling parallel, quantitative analysis of editing outcomes. |
| Cell Synchronization Agents (e.g., Nocodazole, Aphidicolin) | Used to arrest cells in specific cell cycle phases (e.g., S/G2) where HDR is more active, thereby increasing HDR efficiency. |
The ongoing revolution in gene editing, spearheaded by CRISPR-Cas9 technology, is fundamentally rooted in an understanding of bacterial adaptive immune systems. The core thesis underpinning this guide posits that the safety challenges of CRISPR-based therapeutics—namely genomic instability and immunogenicity—are direct consequences of its prokaryotic origins. The Cas9 nuclease, derived from Streptococcus pyogenes, and the system's inherent mechanism of generating double-strand breaks (DSBs) represent a "foreign" biological conflict apparatus repurposed for precise human genome engineering. This phylogenetic disconnect necessitates rigorous validation protocols to assess unintended, host-specific consequences, framing safety evaluation not merely as a regulatory step but as an essential inquiry into the evolutionary compatibility of a bacterial defense system within the mammalian cellular milieu.
Assessing Genomic Instability: Off-Target Effects & Large Rearrangements
Genomic instability arises primarily from off-target editing and on-target genomic rearrangements. Validation requires a multi-faceted approach.
2.1 Key Experimental Protocols
In silico Prediction & GUIDE-seq:
- Protocol: Design sgRNAs using tools like CRISPRseek. For experimental validation, transfert cells with the RNP complex (Cas9 protein + sgRNA) alongside a double-stranded oligodeoxynucleotide (dsODN) "tag." After 72 hours, harvest genomic DNA, shear it, and perform tag-specific enrichment. Prepare sequencing libraries (e.g., for Illumina) and sequence. Bioinformatic analysis identifies off-target sites by mapping tagged integration events.
- Purpose: Genome-wide, unbiased identification of off-target cleavage sites.
CIRCLE-seq (Circularization for *In vitro Reporting of Cleavage Effects by Sequencing):*
- Protocol: Isolate genomic DNA and shear it. End-repair and circularize the fragments using splint adapters under dilute conditions to favor self-ligation. Digest linear DNA with exonuclease, leaving only circular DNA. In vitro, incubate the circularized genomic DNA library with the Cas9:sgRNA RNP complex. Cleaved circles are linearized, then adapter-ligated and PCR-amplified for high-throughput sequencing.
- Purpose: Ultra-sensitive, cell-free profiling of off-target sequences.
Digital Droplet PCR (ddPCR) for Large Deletions & Rearrangements:
- Protocol: Design multiple TaqMan probe assays flanking the on-target cut site and at increasing distances (e.g., 200 bp, 1 kb, 10 kb, 100 kb). Following editing, quantify the copy number of each amplicon relative to a reference locus on a different chromosome. A significant reduction in copy number for distant amplicons indicates a large deletion or rearrangement event.
- Purpose: Quantifying the frequency of large, on-target genomic alterations.
2.2 Quantitative Data Summary
Table 1: Representative Off-Target Analysis Outcomes for a Model Locus (HBB)
| Validation Method | Predicted Top Off-Target Sites | Measured Indel Frequency (%) | Detection Limit |
|---|---|---|---|
| GUIDE-seq (in vivo) | Site 1 (Chr 11), Site 2 (Chr 17) | 0.8%, 0.2% | ~0.01% |
| CIRCLE-seq (in vitro) | 5 additional low-homology sites | Not quantified (in vitro) | ~0.0001% |
| Targeted Amplicon Seq | On-target (HBB) | 85.5% | ~0.1% |
Table 2: Frequency of On-Target Genomic Rearrangements
| Cell Type | Edit Type | ddPCR Amplicon Distance | Frequency of Loss (%) |
|---|---|---|---|
| iPSCs | Knock-in (2 kb donor) | 1 kb flank | 15% |
| iPSCs | Knock-in (2 kb donor) | 10 kb flank | 4% |
| T-cells | Knock-out (RNP) | 5 kb flank | <1% |
2.3 Visualization: Genomic Instability Assessment Workflow
Title: Workflow for Genomic Instability Assessment
Assessing Immunogenicity: Cellular & Humoral Responses
Immunogenicity stems from pre-existing or therapy-induced immune responses to the bacterial-derived Cas9 protein.
3.1 Key Experimental Protocols
Pre-existing Anti-Cas9 Antibody ELISA:
- Protocol: Coat a 96-well plate with purified Cas9 protein (e.g., SpCas9). Incubate with serial dilutions of human serum/plasma from subjects. Detect bound IgG/IgA/IgM antibodies using enzyme-conjugated anti-human secondary antibodies and a colorimetric substrate. Compare absorbance to a standard curve of positive control antibody.
- Purpose: Quantify pre-existing humoral immunity from prior bacterial (e.g., S. pyogenes, S. aureus) exposures.
Cas9-Specific T-cell Activation Assay (ELISpot/Intracellular Cytokine Staining):
- Protocol: Isolate PBMCs from subjects. Stimulate cells in vitro with overlapping peptide pools spanning the Cas9 protein. For ELISpot, capture secreted IFN-γ on a membrane. For ICS, treat cells with a protein transport inhibitor, stain for surface markers (CD4, CD8) and intracellular cytokines (IFN-γ, IL-2), and analyze by flow cytometry.
- Purpose: Detect pre-existing or induced Cas9-specific cellular immune responses.
In vivo Immunogenicity Study (Animal Model):
- Protocol: Administer CRISPR-Cas9 components (e.g., via AAV, LNP) to immunocompetent animal models. Collect sera pre- and post-injection at multiple timepoints for anti-Cas9 antibody ELISA. Harvest splenocytes post-mortem for T-cell activation assays. Analyze tissues for immune infiltrates (histopathology).
- Purpose: Model the de novo immune response to the editing machinery.
3.2 Quantitative Data Summary
Table 3: Representative Immunogenicity Profile Data
| Assay | Population / Model | Positive Result Frequency | Key Metric |
|---|---|---|---|
| Anti-SpCas9 IgG ELISA | Healthy Human Donors (n=200) | ~58% | Median titer: 1:450 |
| Anti-SaCas9 IgG ELISA | Healthy Human Donors (n=200) | ~78% | Median titer: 1:1200 |
| Cas9 T-cell ELISpot (IFN-γ) | In vivo Mouse Study (LNP delivery) | 4/5 mice | >50 SFU/10^6 splenocytes |
| Neutralizing Antibody Assay | Serum from ELISA+ Donors | ~40% of ELISA+ | >50% inhibition of editing |
3.3 Visualization: Anti-Cas9 Immune Response Pathways
Title: Cellular & Humoral Immune Response to Cas9
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagent Solutions for Safety Validation
| Reagent / Material | Function in Validation | Example / Note |
|---|---|---|
| Recombinant Cas9 Proteins | Positive control for immunoassays; component for in vitro cleavage assays (CIRCLE-seq). | HiFi SpCas9, SaCas9; ensure >95% purity. |
| Overlapping Cas9 Peptide Pools | Stimulate Cas9-specific T-cells for ELISpot/ICS assays. | 15-mer peptides, 11-aa overlap, spanning full protein. |
| dsODN "Tag" for GUIDE-seq | Integrates at DSB sites to mark off-target loci for sequencing. | Phosphorothioate-modified ends, HPLC-purified. |
| Digital Droplet PCR (ddPCR) Supermix | Enables absolute quantification of copy number variants for large rearrangement analysis. | Must be optimized for large amplicon detection. |
| Anti-Cas9 Monoclonal Antibody | Critical standard for ELISA assay development and quantification. | Enables generation of a standard curve. |
| CRISPR-Cas9 Edited Reference Cell Lines | Controls for on/off-target sequencing and immunogenicity assays. | Well-characterized clones with known indel profiles. |
| Next-Generation Sequencing Kits | Library prep for GUIDE-seq, CIRCLE-seq, and targeted amplicon sequencing. | Select kits compatible with low-input DNA. |
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) proteins constitute an adaptive immune system in bacteria and archaea. This system provides sequence-specific defense against invasive genetic elements, such as bacteriophages and plasmids. The core thesis of modern genome engineering research is built upon understanding and repurposing these molecular mechanisms, which evolved over millions of years to recognize and cleave foreign nucleic acids. Cas9 and Cas12a are two of the most well-characterized and utilized effector proteins, each representing distinct subtypes (Class 2, Type II and Type V, respectively) with unique biochemical properties that have been harnessed for programmable genome editing, diagnostics, and transcriptional regulation.
Comparative Analysis of Cas9 and Cas12a (Cpf1)
Molecular Architecture and Mechanism
Cas9: A multi-domain protein comprising REC lobes for recognition, a PAM-interacting domain, and HNH and RuvC-like nuclease domains. It requires two RNA molecules: the CRISPR RNA (crRNA) and a trans-activating crRNA (tracrRNA), which are often fused into a single guide RNA (sgRNA). Cas9 creates a blunt-ended double-strand break (DSB) 3 base pairs upstream of the PAM (typically 5'-NGG-3' for Streptococcus pyogenes Cas9).
Cas12a (Cpf1): A single RuvC-like nuclease domain protein that processes its own precursor crRNA (pre-crRNA) and requires only a crRNA for targeting. It recognizes a T-rich PAM (5'-TTTV-3') and creates a double-strand break with staggered ends, leaving a 5' overhang. This enzyme also exhibits collateral, non-specific single-stranded DNA (ssDNA) cleavage activity in trans upon target recognition, a feature exploited in diagnostic applications.
Quantitative Comparison Table
The following table summarizes key characteristics of S. pyogenes Cas9 (SpCas9) and Acidaminococcus Cas12a (AsCas12a).
Table 1: Comparative Properties of SpCas9 and AsCas12a
| Property | Cas9 (SpCas9) | Cas12a (AsCas12a) |
|---|---|---|
| Class/Type | Class 2, Type II | Class 2, Type V |
| Molecular Size | ~1368 amino acids, ~160 kDa | ~1307 amino acids, ~150 kDa |
| Guide RNA | crRNA + tracrRNA (or fused sgRNA) | Single crRNA only |
| crRNA Processing | Requires host RNase III or synthetic sgRNA | Self-processes pre-crRNA |
| PAM Sequence | 5'-NGG-3' (canonical) | 5'-TTTV-3' (where V is A, C, or G) |
| PAM Location | 3' of target sequence (downstream) | 5' of target sequence (upstream) |
| Cleavage Pattern | Blunt-ended DSB | Staggered DSB (5' overhang) |
| Cleavage Site | 3 bp upstream of PAM | 18-23 bp downstream of PAM |
| Nuclease Domains | HNH (cuts target strand), RuvC (cuts non-target) | Single RuvC domain (cuts both strands) |
| Collateral Activity | No | Yes (ssDNA cleavage in trans) |
| Typical Editing Outcome | NHEJ, HDR (blunt ends) | NHEJ, HDR (sticky ends may favor microhomology-mediated repair) |
Other Notable Cas Variants and Their Applications
Beyond Cas9 and Cas12a, the CRISPR toolkit has expanded to include numerous variants with specialized properties.
Cas13 (Type VI): RNA-targeting effector with RNAse activity and robust collateral cleavage of bystander RNA, enabling sensitive nucleic acid detection (e.g., SHERLOCK).
Cas12f (Cas14, Type V-F): Ultra-small (~400-700 aa) nucleases derived from archaea, enabling delivery via compact viral vectors like AAV.
CasΦ (Cas12j, Type V-U): A hypercompact Cas protein (~70-80 kDa) from huge phages with a single active site for DNA cleavage.
Base Editors: Fusions of catalytically impaired Cas9/Cas12a with deaminase enzymes (e.g., cytidine or adenosine deaminase) enabling direct, template-free conversion of single nucleotides without creating a DSB.
Prime Editors: A fusion of Cas9 nickase with a reverse transcriptase, programmed with a prime editing guide RNA (pegRNA) to perform precise insertions, deletions, and all base-to-base conversions with minimal byproducts.
Table 2: Overview of Additional CRISPR-Cas Systems
| Variant | Class/Type | Target | Key Feature | Primary Application |
|---|---|---|---|---|
| Cas13a | Class 2, Type VI | RNA | Collateral RNAse activity | RNA detection, knockdown, editing |
| Cas12f | Class 2, Type V-F | DNA | Ultra-small size (<500 aa) | Delivery-constrained settings (e.g., AAV) |
| CasΦ | Class 2, Type V-U | DNA | Compact, single active site | Basic research, potential for delivery |
| BE4max | Fusion (dCas9) | DNA | Cytosine base editor; high efficiency & purity | Base substitution (C•G to T•A) |
| PE2 | Fusion (nCas9-RT) | DNA | Reverse transcriptase-mediated template writing | Precise small edits without DSB |
Experimental Protocols for Key Functional Assays
Protocol:In VitroPAM Depletion Assay for PAM Identification
Purpose: To empirically determine the PAM sequence recognized by a novel or engineered Cas nuclease. Reagents: Purified Cas protein, genomic DNA from a neutral organism (e.g., lambda phage), in vitro transcription kit, DNase I, NGS library prep kit.
- Library Preparation: Digest genomic DNA with DNase I to generate random fragments. Ligate to a common adaptor sequence.
- Targeted Cleavage: Incubate the DNA library with the Cas protein and a pool of crRNAs containing random spacer sequences.
- Size Selection: Run reaction products on an agarose gel. Isulate the cleaved, shorter fragments.
- Sequencing & Analysis: Prepare an NGS library from the isolated fragments. Sequence and align reads to the original genome. The sequence immediately adjacent to the cleavage site (consistent across all cleaved fragments) represents the functional PAM.
Protocol: Cell-Based Editing Efficiency and Specificity Assay
Purpose: To quantify on-target editing efficiency and assess off-target effects of a Cas-gRNA complex in mammalian cells. Reagents: HEK293T cells, Lipofectamine 3000, plasmid expressing Cas protein and gRNA, genomic DNA extraction kit, T7 Endonuclease I (T7EI) or Surveyor nuclease, NGS-based off-target prediction software, primers for on-/off-target loci.
- Transfection: Co-transfect HEK293T cells with the Cas expression plasmid and a plasmid expressing the specific gRNA.
- Harvest Genomic DNA: 72 hours post-transfection, extract genomic DNA from harvested cells.
- On-target Analysis (T7EI Assay): PCR-amplify the genomic region surrounding the on-target site. Denature and reanneal the PCR products to form heteroduplexes if indels are present. Digest with T7EI, which cleaves mismatched DNA. Analyze fragments by gel electrophoresis; band intensity correlates with editing efficiency.
- Off-target Analysis (NGS): Using computational prediction (e.g., Cas-OFFinder), identify potential off-target sites. Amplify these loci from the genomic DNA by PCR and subject them to deep sequencing. Align reads to the reference genome to detect insertion/deletion mutations at frequencies above background.
Visualization of Core Mechanisms and Workflows
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-Cas Research
| Reagent / Material | Function / Application |
|---|---|
| High-Fidelity Cas9/Cas12a Expression Plasmid | Ensures reliable, high-level expression of the nuclease in mammalian, bacterial, or other cell types with appropriate promoters and nuclear localization signals (NLS). |
| sgRNA/crRNA Cloning Vector | Backbone plasmid for efficient synthesis and expression of guide RNA sequences, often containing a U6 or T7 promoter. |
| In Vitro Transcription Kit (T7) | For producing high-yield, pure gRNA, crRNA, and tracrRNA for in vitro assays or RNP delivery. |
| Recombinant Purified Cas Protein | For biochemical assays (PAM depletion, in vitro cleavage), structural studies, and direct RNP delivery into cells. |
| T7 Endonuclease I (T7EI) | Mismatch-specific endonuclease used in the Surveyor/T7EI assay to detect and quantify indel mutations at target loci. |
| NGS-Based Off-Target Analysis Kit | Commercial kits (e.g., Illumina, IDT) for preparing sequencing libraries from amplified genomic loci to detect low-frequency off-target edits. |
| Electroporation or Lipofection Reagent | For efficient delivery of CRISPR components (plasmids, RNPs) into hard-to-transfect cell lines or primary cells. |
| Validated Positive Control gRNA | A guide RNA with known high editing efficiency (e.g., targeting the AAVS1 safe harbor locus in human cells) to control for experimental workflow integrity. |
| Fluorescent ssDNA Reporter (for Cas12a/13) | A quenched fluorescent oligonucleotide that is cleaved upon Cas12a/Cas13 collateral activity, enabling real-time detection of target recognition (used in DETECTR, SHERLOCK). |
| HDR Donor Template | Single-stranded oligodeoxynucleotide (ssODN) or double-stranded DNA (dsDNA) template containing the desired edit, used to guide Homology-Directed Repair (HDR) for precise gene correction or insertion. |
Research into the evolutionary origins of the CRISPR-Cas9 bacterial immune system necessitates rigorous validation across biological hierarchies. This journey from molecular mechanism to physiological function requires a cascade of model systems, each with increasing complexity. Validation in cell lines establishes molecular causality, organoids introduce tissue-specific architecture, and animal models confirm systemic functionality. This guide details the technical frameworks for this validation cascade within CRISPR research.
Cell Lines: Establishing Molecular Causality
Cell lines provide a homogenous, genetically tractable system for initial validation of CRISPR-related components and mechanisms.
Key Experimental Protocol: Validating Anti-Phage Activity in a Bacterial Cell Line
- Objective: To confirm the functional immune response of a reconstructed ancestral CRISPR-Cas system against bacteriophage infection.
- Materials: An engineered E. coli strain lacking its native CRISPR systems; plasmid vectors encoding putative ancestral Cas genes and a synthetic CRISPR array; target bacteriophage (e.g., λ phage).
- Method:
- Reconstitution: Co-transform the engineered E. coli with the Cas expression plasmid and the CRISPR array plasmid targeting a conserved essential gene of the λ phage.
- Challenge Assay: Grow transformed bacteria to mid-log phase. Incubate with serial dilutions of λ phage at a low multiplicity of infection (MOI ~0.1).
- Plaque Assay: After lysis period, mix culture with soft agar and a lawn of susceptible indicator bacteria. Pour onto agar plates.
- Quantification: Incubate and count plaque-forming units (PFUs). Compare PFU counts from bacteria expressing the CRISPR system versus empty vector controls.
- Data Interpretation: A statistically significant reduction in plaque count indicates functional immune activity. Sequencing surviving phage can reveal escape mutations, validating target specificity.
Table 1: Quantitative Output of CRISPR Anti-Phage Validation in E. coli
| Experimental Condition | Avg. Plaque Count (PFU/mL) | Standard Deviation | % Reduction vs Control | p-value |
|---|---|---|---|---|
| Control (Empty Vector) | 2.5 x 10^8 | 3.1 x 10^7 | 0% | N/A |
| Ancestral Cas9 System | 1.2 x 10^6 | 2.5 x 10^5 | 99.5% | <0.001 |
| Spacer-Deletion Mutant | 2.4 x 10^8 | 2.8 x 10^7 | 4.0% | 0.35 |
Organoids: Introducing Tissue Context
Mammalian intestinal or stem cell organoids model complex cellular environments, allowing validation of CRISPR systems in eukaryotic cells and tissue-like structures.
Key Experimental Protocol: Assessing Off-Target Effects in Human Colon Organoids
- Objective: To evaluate the specificity of a Cas9 variant (e.g., high-fidelity Cas9-HF1) derived from evolutionary studies when targeting a disease-relevant locus.
- Materials: Human intestinal stem cell-derived organoids; nucleofection reagents; RNP complexes (Cas9-HF1 protein + sgRNA); next-generation sequencing (NGS) library prep kits.
- Method:
- Editing: Dissociate organoids to single cells. Nucleofect cells with RNP complexes targeting the APC gene.
- Recovery & Expansion: Culture cells to regenerate edited organoids over 7-10 days.
- On-Target Analysis: Sanger sequence the targeted APC locus from bulk organoid DNA to calculate indel efficiency.
- Off-Target Analysis: Perform NGS on predicted top 10 off-target sites (from in silico tools like GUIDE-seq) and whole-exome sequencing on edited and control organoid lines.
- Data Interpretation: High on-target editing with minimal indels at predicted off-target sites and no significant increase in exome-wide variants validates high specificity.
Table 2: NGS Analysis of CRISPR-Cas9-HF1 Editing in Intestinal Organoids
| Genomic Locus | Read Depth | % Indels (Wild-Type Cas9) | % Indels (Cas9-HF1) | Predicted Mismatch Tolerance |
|---|---|---|---|---|
| On-Target: APC Exon 15 | 12,000 | 78.5% | 72.1% | N/A |
| Off-Target 1 (3 mismatches) | 10,500 | 5.2% | 0.15% | 3 |
| Off-Target 2 (2 mismatches) | 11,800 | 12.7% | 0.08% | 2 |
| Off-Target 3 (4 mismatches) | 9,500 | 0.8% | 0.01% | 4 |
Animal Models: Systemic Functional Validation
Transgenic animal models (e.g., mice, zebrafish) provide the final validation tier, assessing CRISPR system function, delivery, and immune responses in vivo.
Key Experimental Protocol: In Vivo Efficacy of a CRISPR-Based Antimicrobial
- Objective: To test an engineered CRISPR-Cas system that targets antibiotic-resistant genes in a mouse model of Staphylococcus aureus infection.
- Materials: BALB/c mice; bioluminescent S. aureus strain harboring a plasmid with a resistance gene (mecA); cationic lipid nanoparticles (LNPs) loaded with plasmid DNA expressing Cas9 and specific sgRNA; IVIS imaging system.
- Method:
- Infection: Establish a localized subcutaneous infection with bioluminescent S. aureus.
- Treatment: Administer CRISPR-LNP formulations via intraperitoneal or local injection at 24h post-infection. Controls include empty LNPs and conventional antibiotics.
- Monitoring: Track infection burden via bioluminescence imaging daily for 5 days.
- Endpoint Analysis: Harvest tissue at endpoint for bacterial load (CFU counts), PCR analysis of mecA gene disruption, and histopathology for inflammation.
- Data Interpretation: A significant reduction in bioluminescence and CFU in the CRISPR group, coupled with genomic disruption of mecA, validates in vivo efficacy and potential therapeutic application.
Table 3: In Vivo Efficacy of CRISPR Antimicrobial in Mouse Infection Model
| Treatment Group (n=8) | Day 3 Avg. Bioluminescence (p/s/cm²/sr) | Day 5 Avg. CFU/g Tissue | % mecA Disruption in Recovered Bacteria |
|---|---|---|---|
| Untreated Control | 3.2 x 10^5 | 1.8 x 10^8 | 0% |
| Vancomycin (Positive Control) | 8.4 x 10^4 | 5.5 x 10^5 | 0% |
| CRISPR-LNP (mecA target) | 1.1 x 10^4 | 9.2 x 10^4 | 67% |
| CRISPR-LNP (Scrambled sgRNA) | 2.9 x 10^5 | 1.4 x 10^8 | <1% |
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Validation |
|---|---|
| Engineered "CRISPR-Null" Bacterial Strains | Provide a clean background for reconstituting and testing putative ancestral CRISPR systems without interference from native machinery. |
| Recombinant Cas Protein (Wild-type & Variants) | For forming RNP complexes in eukaryotic cells, allowing rapid editing and reducing plasmid-based cytotoxicity. |
| Synthetic sgRNA with Chemical Modifications | Enhances stability and reduces immunogenicity in mammalian cells and in vivo applications. |
| Stem Cell-Derived Organoid Culture Kits | Provides standardized matrices and media for robust generation of tissue-specific organoids for editing studies. |
| Cationic Lipid Nanoparticles (LNPs) | Enables efficient in vivo delivery of CRISPR payloads (RNA or DNA) to target tissues. |
| In Vivo Imaging Systems (e.g., IVIS) | Allows longitudinal, non-invasive tracking of disease progression (e.g., infection, cancer) and therapeutic efficacy in live animals. |
| Off-Target Prediction & Validation Suites | Software (e.g., Cas-OFFinder) and NGS kits (e.g., GUIDE-seq, CIRCLE-seq) for comprehensive specificity profiling. |
Visualizations
Diagram 1: Validation Cascade from Molecules to Organisms
Diagram 2: Protocol for Validating Anti-Phage Activity
Diagram 3: Key Pathways in CRISPR-Cas9 Immune Function
Conclusion
The journey of CRISPR-Cas9 from a bacterial immune system to a transformative biomedical tool exemplifies the power of fundamental biological discovery. This article has synthesized its foundational origins, methodological adaptations, critical optimization challenges, and validated performance relative to other technologies. For researchers and drug developers, the key takeaway is that the system's simplicity, versatility, and continual refinement through protein engineering offer an unparalleled platform for probing biology and developing next-generation therapies. Future directions hinge on solving delivery and specificity challenges at a clinical scale, expanding the editing toolbox (e.g., base, prime, and epigenome editors), and navigating the evolving ethical and regulatory landscape. Ultimately, understanding its prokaryotic roots is essential for innovating its eukaryotic applications, promising a new era of precise genetic medicine.