Mastering ChIP-seq: A Comprehensive Protocol Guide for Epigenetic Discovery and Drug Development
This detailed guide provides researchers and drug development professionals with a complete roadmap for successful ChIP-seq experiments.
Mastering ChIP-seq: A Comprehensive Protocol Guide for Epigenetic Discovery and Drug Development
Abstract
This detailed guide provides researchers and drug development professionals with a complete roadmap for successful ChIP-seq experiments. We cover the foundational principles of chromatin immunoprecipitation followed by sequencing, from core concepts and antibody selection to a step-by-step optimized protocol. The article delves into critical troubleshooting for common pitfalls, advanced optimization strategies for challenging samples, and rigorous validation methods to ensure data integrity. Finally, we compare ChIP-seq with emerging techniques like CUT&Tag and ATAC-seq, offering insights for experimental design. This resource empowers scientists to generate high-quality, reproducible epigenomic data to drive discoveries in gene regulation, disease mechanisms, and therapeutic target identification.
ChIP-seq Fundamentals: Decoding the Epigenomic Landscape from First Principles
Introduction to Epigenetics and the Power of Protein-DNA Interaction Mapping
1. Introduction and Context Epigenetics refers to heritable changes in gene expression that do not involve alterations to the underlying DNA sequence. These changes, including DNA methylation, histone modifications, and chromatin remodeling, constitute a critical regulatory layer. Mapping the precise genomic locations where proteins, such as transcription factors or modified histones, interact with DNA is fundamental to decoding the epigenome. Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) has emerged as the cornerstone protocol for generating high-resolution maps of protein-DNA interactions, driving hypothesis generation in basic research and target validation in drug development.
2. Quantitative Data Summary: Key Epigenetic Marks and Outcomes
Table 1: Common Histone Modifications and Their Functional Associations
| Histone Mark | Typical Genomic Association | General Functional Outcome | Relevance to Disease/Drug Discovery |
|---|---|---|---|
| H3K4me3 | Promoters of active genes | Transcriptional activation | Altered in cancers; target for epigenetic therapy. |
| H3K27ac | Active enhancers and promoters | Enhancer/promoter activity | Defines super-enhancers in oncology. |
| H3K27me3 | Promoters of silenced genes | Transcriptional repression (Polycomb) | Misregulated in developmental disorders & cancer. |
| H3K9me3 | Heterochromatin, repetitive elements | Transcriptional silencing | Genome instability marker. |
| H3K36me3 | Gene bodies of transcribed genes | Elongation-associated, splicing | Correlates with mutation rates in cancer. |
Table 2: Comparative Overview of Key Protein-DNA Mapping Technologies
| Method | Target | Resolution | Throughput | Primary Application in Epigenomics |
|---|---|---|---|---|
| ChIP-seq | Protein-DNA interactions | ~100-300 bp | Moderate | Genome-wide mapping of TF binding & histone marks. |
| CUT&RUN | Protein-DNA interactions | ~10-50 bp (in situ) | High | Low-cell-number, high-resolution mapping. |
| ATAC-seq | Chromatin accessibility | ~1 bp (insert size) | High | Mapping open chromatin regions & nucleosome position. |
| Hi-ChIP | Protein-anchored chromatin loops | ~1-5 kb (contact) | Moderate | Mapping long-range interactions linked to a specific protein. |
3. Detailed Protocol: Standard Crosslinking ChIP-seq for Histone Modifications
Application Note: This protocol is optimized for generating genome-wide maps of histone modifications (e.g., H3K27ac) from mammalian cell lines, a critical step in identifying active regulatory elements.
Materials & Reagents:
- Formaldehyde (37%): For crosslinking protein to DNA.
- Glycine (2.5M): To quench crosslinking.
- Lysis Buffers: Cell Lysis Buffer & Nuclear Lysis Buffer (containing SDS).
- Micrococcal Nuclease (MNase) or Sonication Device: For chromatin shearing.
- Protein A/G Magnetic Beads: For antibody-antigen complex capture.
- Validated Antibody: Specific to the histone mark of interest (e.g., anti-H3K27ac).
- ChIP Elution Buffer: (1% SDS, 0.1M NaHCO3).
- DNA Clean-up Kit: For purifying immunoprecipitated DNA.
- High-Sensitivity DNA Assay Kit: For quantifying library DNA.
Procedure:
- Crosslinking: Add 37% formaldehyde directly to cell culture medium (final concentration 1%). Incubate 10 min at room temperature. Quench with 2.5M glycine.
- Cell Lysis & Chromatin Preparation: Wash cells. Resuspend pellet in Cell Lysis Buffer. Pellet nuclei. Lyse nuclei in Nuclear Lysis Buffer.
- Chromatin Shearing: Using a focused ultrasonicator, shear chromatin to an average size of 200-500 bp. Alternative for histones: Use MNase digestion for nucleosome-level resolution.
- Immunoprecipitation: Pre-clear sheared chromatin with beads. Incubate chromatin with target-specific antibody overnight at 4°C. Add Protein A/G beads and incubate to capture complexes.
- Washes & Elution: Wash beads sequentially with Low Salt, High Salt, LiCl, and TE buffers. Elute complexes in ChIP Elution Buffer. Reverse crosslinks at 65°C with NaCl.
- DNA Purification: Treat with Proteinase K and RNase A. Purify DNA using a spin column kit.
- Library Preparation & Sequencing: Use the purified DNA to construct a sequencing library (end repair, A-tailing, adapter ligation, PCR amplification). Validate library quality and sequence on an appropriate platform.
4. The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for ChIP-seq and Epigenetic Analysis
| Item | Function & Importance |
|---|---|
| Validated ChIP-grade Antibodies | Specificity is paramount; non-specific antibodies lead to high background and false peaks. |
| Magnetic Beads (Protein A/G) | Enable efficient pull-down and easy washing of antibody complexes, reducing background. |
| High-Fidelity DNA Polymerase | For accurate, unbiased amplification of low-input ChIP DNA during library prep. |
| Dual-Indexed Adapters | Allow multiplexing of many samples in a single sequencing run, reducing cost. |
| Size Selection Beads | Critical for selecting optimally sized DNA fragments post-sonication and post-library prep. |
| Cell Permeable Histone Deacetylase (HDAC) Inhibitors | Tool compounds to manipulate the epigenome (e.g., TSA) and validate ChIP targets. |
| Next-Generation Sequencing Kit | Platform-specific chemistry for final cluster generation and sequencing. |
5. Visualized Workflows and Pathways
Diagram Title: Standard ChIP-seq Experimental Workflow
Diagram Title: Histone Modification Signaling Pathway
Within the context of a broader thesis on the ChIP-seq protocol for epigenomics research, understanding the core immunoprecipitation mechanism is fundamental. Chromatin Immunoprecipitation (ChIP) is the pivotal technique that enables the selective isolation of DNA sequences bound by specific proteins in their native chromatin context. This capture is the critical first step before sequencing (ChIP-seq), allowing researchers to map protein-DNA interactions genome-wide, which is essential for elucidating gene regulatory networks in development, disease, and drug response.
Core Mechanism: The Principle of Capture
The ChIP process isolates protein-bound DNA through a series of steps that preserve in vivo interactions. The central mechanism relies on the specificity of antibody-antigen recognition to precipitate a protein of interest along with its crosslinked DNA fragments.
- Crosslinking: Live cells or tissues are treated with formaldehyde, creating covalent bonds between proteins and DNA that are in close spatial proximity. This "freezes" transient interactions.
- Chromatin Fragmentation: The crosslinked chromatin is physically sheared (typically via sonication or enzymatic digestion) into small fragments (200-1000 bp). This solubilizes the chromatin and renders it accessible for immunoprecipitation.
- Immunoprecipitation: The fragmented chromatin is incubated with a bead-conjugated antibody highly specific to the protein of interest (e.g., a transcription factor, histone modification, or polymerase). The antibody-bead complex binds to the target protein, and through it, captures the crosslinked DNA fragment.
- Washing & Elution: Beads are washed stringently to remove non-specifically bound chromatin. The crosslinks are then reversed (typically by heating), releasing the captured DNA from the protein-antibody-bead complex.
- Purification: The released DNA is purified, resulting in a sample enriched for genomic regions that were bound by the protein of interest.
Table 1: Key Quantitative Parameters for Standard ChIP Protocol
| Parameter | Typical Range/Value | Importance & Impact |
|---|---|---|
| Formaldehyde Concentration | 0.5 - 1.5% | Higher % increases crosslinking efficiency but reduces chromatin shearing efficiency and antigen accessibility. |
| Crosslinking Time | 5 - 30 minutes | Longer times stabilize weak interactions but can increase epitope masking. |
| Sonication Fragment Size | 200 - 500 bp (for transcription factors) | Smaller fragments give higher resolution mapping. Affects signal-to-noise in sequencing. |
| Chromatin Input per IP | 1 - 10 µg | Must be optimized based on target abundance. Low abundance targets require more input. |
| Antibody Amount per IP | 1 - 10 µg | Insufficient antibody reduces yield; excess increases non-specific binding. |
| Wash Stringency (Salt Conc.) | 150 - 500 mM NaCl | Higher salt reduces non-specific ionic interactions but may disrupt weak specific interactions. |
| DNA Yield after Purification | 1 - 100 ng | Highly variable; depends on target abundance, antibody quality, and cell number. Low yield is a major challenge for low-abundance factors. |
Detailed Protocol: Crosslinking & Sonication ChIP for Cultured Cells
Materials & Reagents
- Cell culture
- 37% Formaldehyde
- 2.5M Glycine (in PBS)
- PBS, ice-cold
- Cell Lysis Buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% NP-40, protease inhibitors)
- Nuclei Lysis/Sonication Buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS, protease inhibitors)
- Dilution Buffer (16.7 mM Tris-HCl pH 8.0, 167 mM NaCl, 1.2 mM EDTA, 1.1% Triton X-100, 0.01% SDS)
- Protein A/G Magnetic Beads
- Specific antibody and isotype control IgG
- Low Salt Wash Buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS)
- High Salt Wash Buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS)
- LiCl Wash Buffer (10 mM Tris-HCl pH 8.0, 250 mM LiCl, 1 mM EDTA, 1% NP-40, 1% sodium deoxycholate)
- TE Buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA)
- Elution Buffer (1% SDS, 100 mM NaHCO3)
- Proteinase K, RNase A
- Phenol:Chloroform:Isoamyl alcohol, Ethanol, Glycogen
Method
- Crosslinking: For adherent cells, add 37% formaldehyde directly to culture medium to a final concentration of 1%. Incubate for 10 minutes at room temperature with gentle rocking.
- Quenching: Add 2.5M glycine to a final concentration of 0.125M. Incubate for 5 minutes to stop crosslinking.
- Cell Harvesting: Aspirate medium, wash cells twice with ice-cold PBS. Scrape cells into PBS, pellet by centrifugation (500 x g, 5 min, 4°C).
- Cell Lysis: Resuspend pellet in 1 mL Cell Lysis Buffer. Incubate on ice for 15 minutes. Pellet nuclei (2,000 x g, 5 min, 4°C).
- Nuclei Lysis: Resuspend pellet in 500 µL Nuclei Lysis/Sonication Buffer. Incubate on ice for 10 minutes.
- Chromatin Shearing (Sonication):
- Transfer lysate to a microTUBE. Shear using a focused ultrasonicator (e.g., Covaris) or bath sonicator.
- Optimized Settings (Covaris S2): Duty Cycle: 5%, Intensity: 4, Cycles per Burst: 200, Time: 10-15 minutes (to achieve 200-500 bp fragments).
- Pellet debris (16,000 x g, 10 min, 4°C). Transfer supernatant (sheared chromatin) to a new tube.
- Immunoprecipitation:
- Dilute sheared chromatin 10-fold with Dilution Buffer.
- Pre-clear with 20 µL Protein A/G beads for 1 hour at 4°C.
- Take an "Input" sample (2%). To the rest, add specific antibody (e.g., 5 µg) and incubate overnight at 4°C with rotation.
- Bead Capture & Washes:
- Add 40 µL pre-blocked Protein A/G beads and incubate for 2 hours.
- Pellet beads and wash sequentially for 5 minutes each with rotation: 1x Low Salt Buffer, 1x High Salt Buffer, 1x LiCl Buffer, 2x TE Buffer.
- Elution & Decrosslinking:
- Elute chromatin from beads with 200 µL Elution Buffer by vortexing for 15 minutes at room temperature.
- Add 8 µL 5M NaCl to eluates and the saved Input sample. Heat at 65°C for 4-6 hours (or overnight) to reverse crosslinks.
- DNA Purification:
- Add 10 µL 0.5M EDTA, 20 µL 1M Tris-HCl pH 6.5, and 2 µL Proteinase K (20 mg/mL). Incubate at 45°C for 2 hours.
- Purify DNA by phenol-chloroform extraction and ethanol precipitation with glycogen carrier.
- Resuspend DNA in TE buffer or nuclease-free water. The captured DNA is now ready for qPCR analysis or library preparation for sequencing.
Visualizing the Core ChIP Mechanism
ChIP Workflow to Capture Protein-Bound DNA
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Effective ChIP
| Reagent | Function & Critical Role in Capture Mechanism |
|---|---|
| High-Quality, ChIP-Validated Antibody | The cornerstone of specificity. Must recognize the target epitope even after crosslinking and denaturation. Poor antibody performance is the leading cause of ChIP failure. |
| Protein A/G Magnetic Beads | Provide a solid support for antibody immobilization and easy separation via magnetism. Reduce non-specific background compared to agarose beads. |
| Formaldehyde (Ultra Pure) | Creates protein-DNA and protein-protein crosslinks, "trapping" transient interactions for capture. Purity is essential for reproducibility. |
| Protease Inhibitor Cocktail (PIC) | Prevents degradation of the target protein and histone epitopes during cell lysis and chromatin preparation, preserving the target for immunoprecipitation. |
| Covaris microTUBE or equivalent | Ensures consistent, efficient, and reproducible chromatin shearing via focused ultrasonication, which is critical for resolution and yield. |
| RNase A & Proteinase K | RNase removes contaminating RNA after elution. Proteinase K digests proteins (including antibodies) after decrosslinking, allowing clean DNA purification. |
| Glycogen (Molecular Biology Grade) | Acts as an inert carrier during ethanol precipitation of low-concentration DNA, dramatically improving recovery of the precious captured DNA. |
| Magnetic Rack | Enables efficient bead separation during wash and elution steps, minimizing physical loss of the bead-bound complex. |
The study of protein-DNA interactions is fundamental to epigenomics. The transition from Chromatin Immunoprecipitation coupled with microarray (ChIP-chip) to next-generation sequencing based ChIP-seq represents a paradigm shift. This application note details modern ChIP-seq protocols within the broader thesis of achieving high-resolution, genome-wide mapping of histone modifications and transcription factor binding sites for drug target discovery.
Table 1: Quantitative Comparison of ChIP-chip vs. ChIP-seq
| Feature | ChIP-chip | Modern ChIP-seq (Illumina NovaSeq) |
|---|---|---|
| Genomic Coverage | Limited to probe regions | Comprehensive, unbiased |
| Resolution | ~100 bp (practical) | <10 bp (theoretical) |
| Dynamic Range | ~2-3 orders of magnitude | >4 orders of magnitude |
| Input DNA Required | High (microgram) | Low (nanogram) |
| Typical Run Time | 3-5 days (hyb + array) | 1-3 days (seq) |
| Cost per Sample (2024) | ~$400 (array only) | ~$200-$500 (seq only) |
| Primary Limitation | Array design, hybridization bias | PCR amplification bias, cost of sequencing |
Detailed ChIP-seq Protocol for Histone Modification Mapping
This protocol is optimized for frozen cell pellets or tissues.
Day 1: Cell Fixation & Chromatin Preparation
- Crosslinking: Resuspend ~1x10^6 cells in 1 mL growth medium. Add 27 µL of 37% formaldehyde (final 1%). Incubate 10 min at room temperature (RT) with rotation.
- Quenching: Add 100 µL of 1.25 M glycine. Incubate 5 min at RT.
- Washing: Pellet cells, wash 2x with cold PBS + protease inhibitors.
- Lysis: Lyse cells in 1 mL Lysis Buffer 1 (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5% NP-40, 0.25% Triton X-100) for 10 min at 4°C. Pellet.
- Nuclear Lysis: Resuspend in 1 mL Lysis Buffer 2 (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) for 10 min at 4°C. Pellet.
- Chromatin Shearing: Resuspend pellet in 300 µL Sonication Buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris-HCl pH 8.0). Sonicate using a Covaris S220 (or equivalent) to achieve 200-500 bp fragments (e.g., 20 cycles: 30 sec ON, 30 sec OFF, high power). Clear supernatant by centrifugation.
- Immunoprecipitation: Dilute sheared chromatin 10-fold in ChIP Dilution Buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH 8.0, 167 mM NaCl). Add 1-5 µg of validated antibody (e.g., anti-H3K27ac). Incubate with rotation overnight at 4°C.
Day 2: Bead Capture & Wash
- Capture: Add 50 µL of pre-blocked Protein A/G magnetic beads. Incubate 2 hours at 4°C.
- Washing: Wash beads sequentially on a magnetic rack with:
- Low Salt Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 150 mM NaCl)
- High Salt Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 500 mM NaCl)
- LiCl Wash Buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH 8.0)
- 2x with TE Buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA)
- Elution: Elute DNA twice with 150 µL Elution Buffer (1% SDS, 100 mM NaHCO3). Combine eluates.
- Reverse Crosslinking: Add NaCl to 200 mM and RNase A. Incubate overnight at 65°C.
Day 3: DNA Purification & Library Preparation
- Proteinase K Digestion: Add Proteinase K, incubate 2 hours at 45°C.
- DNA Purification: Purify DNA using SPRI beads (e.g., AMPure XP). Elute in 30 µL TE.
- Library Construction: Use a commercial library prep kit (e.g., NEB Next Ultra II DNA). Steps include end-repair, A-tailing, adapter ligation, and size selection (target 300-500 bp inserts).
- Library Amplification: Perform 10-12 cycles of PCR with indexed primers. Purify final library.
Day 4: Sequencing
Quantify library by qPCR (for molarity) and fragment analyzer. Pool libraries and sequence on an Illumina platform (e.g., NovaSeq 6000, PE 50 bp). Aim for 20-40 million reads per histone mark sample.
Modern ChIP-seq Experimental Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Robust ChIP-seq
| Item | Function & Critical Note | Example Product/Supplier |
|---|---|---|
| Validated ChIP-grade Antibody | Target-specific immunoprecipitation; the most critical variable. Must be validated for ChIP-seq. | Cell Signaling Tech. (CST), Abcam, Diagenode |
| Magnetic Protein A/G Beads | Efficient capture of antibody-bound complexes; reduce non-specific binding. | Dynabeads (Thermo), SureBeads (Bio-Rad) |
| Covaris Sonicator | Consistent, reproducible chromatin shearing to optimal fragment size. | Covaris S220/E220 |
| SPRI Size Selection Beads | Clean-up and size selection of DNA after elution and during library prep. | AMPure XP (Beckman), SPRIselect |
| NGS Library Prep Kit | Converts low-input ChIP DNA into sequencing-ready libraries with high complexity. | NEB Next Ultra II, Illumina TruSeq ChIP |
| Dual Indexed Adapters | Enables multiplexing of many samples in a single sequencing run. | IDT for Illumina, TruSeq indexes |
| High-Fidelity PCR Mix | Amplifies libraries with minimal bias and errors during indexing PCR. | KAPA HiFi, NEB Q5 |
| Bioanalyzer/TapeStation | QC for sheared chromatin and final library fragment size distribution. | Agilent 2100, 4200 |
Bioinformatic Analysis Pathway
ChIP-seq Data Analysis Pipeline
Advanced Protocol: Low-Input and Single-Cell ChIP-seq (scChIP-seq)
For scarce clinical samples or cellular heterogeneity studies.
Key Modifications:
- Micrococcal Nuclease (MNase) Digestion: Use MNase for fragmentation to maximize epitope availability from low cell counts (<10,000).
- Carrier DNA/RNA: Add inert carrier (e.g., D. melanogaster chromatin) during IP to improve bead capture kinetics.
- Tagmentation-based Library Prep: Use Th5 transposase (e.g., Nextera) for direct "tagmentation" of bead-bound chromatin, minimizing purification losses.
- Cell Barcoding: For scChIP-seq, use droplet-based platforms (e.g., Drop-seq) or combinatorial indexing to barcode individual nuclei before pooling.
Table 3: Comparison of Standard vs. Low-Input ChIP-seq
| Parameter | Standard ChIP-seq | Low-Input/scChIP-seq |
|---|---|---|
| Starting Cell Number | 0.5-1 million | 100 - 10,000 |
| Fragmentation Method | Sonication (Covaris) | MNase Digestion |
| Critical Step | Shearing efficiency | Minimizing sample loss |
| Library Method | Ligation-based | Tagmentation-based |
| Primary Challenge | Background signal | Library complexity |
| Read Depth Required | 20-40 million | 5-10 million (per cell pool) |
Application Notes
ChIP-seq (Chromatin Immunoprecipitation followed by sequencing) is the cornerstone technique for profiling genome-wide protein-DNA interactions. Within epigenomics research, it is indispensable for mapping the binding sites of transcription factors (TFs), the localization of histone modifications, and the identification of regulatory elements such as promoters, enhancers, and silencers. These maps are fundamental for understanding gene regulatory networks in development, disease, and drug response.
Mapping Transcription Factors: ChIP-seq for TFs provides a snapshot of direct DNA binding events, revealing primary regulatory nodes. This is critical for constructing gene regulatory networks and identifying master regulators in cellular differentiation or oncogenesis.
Mapping Histone Modifications: Specific histone post-translational modifications correlate with distinct chromatin states. For example, H3K4me3 marks active promoters, H3K27ac marks active enhancers, and H3K9me3 marks heterochromatin. Profiling these modifications allows for the segmentation of the genome into functional regulatory domains.
Identifying Regulatory Elements: Integrative analysis of TF binding and histone modification maps enables the precise annotation of enhancers, super-enhancers, and other cis-regulatory modules. This is vital for interpreting non-coding genetic variation associated with disease.
Quantitative Data Summary: The following table summarizes key metrics and outcomes from typical ChIP-seq experiments targeting different factors.
Table 1: Typical Outcomes and Metrics for Key ChIP-seq Applications
| Target Class | Example Target | Typical Peak Count | Common Antibody Clonality | Primary Biological Insight |
|---|---|---|---|---|
| Transcription Factor | p53, STAT1 | 10,000 - 50,000 | Monoclonal | Direct DNA binding sites; core regulatory circuits. |
| Histone Modification (Activation) | H3K27ac, H3K4me3 | 50,000 - 200,000+ | Polyclonal | Active promoters and enhancers; regulatory landscape. |
| Histone Modification (Repression) | H3K9me3, H3K27me3 | Large, broad domains | Polyclonal | Silenced genomic regions; facultative/constitutive heterochromatin. |
| Chromatin Regulator | RNA Polymerase II, BRD4 | Varies (e.g., Pol II: 20,000-100,000) | Monoclonal/Polyclonal | Transcriptional activity and elongation; engagement at regulatory elements. |
Detailed Protocols
Protocol 1: Crosslinking ChIP-seq for Transcription Factors
Principle: Reversible crosslinking captures transient TF-DNA interactions.
- Cell Fixation: Treat cells with 1% formaldehyde for 8-10 minutes at room temperature. Quench with 125mM glycine.
- Cell Lysis & Chromatin Shearing: Lyse cells. Isolate nuclei. Sonicate chromatin to 200-500 bp fragments using a focused ultrasonicator. Optimization Tip: Perform a shearing test run and check fragment size by agarose gel electrophoresis.
- Immunoprecipitation: Incubate sheared chromatin with 1-5 µg of validated, high-specificity antibody against the target TF overnight at 4°C with rotation. Use Protein A/G magnetic beads for capture.
- Washes & Elution: Wash beads sequentially with Low Salt, High Salt, LiCl, and TE buffers. Elute bound complexes with fresh elution buffer (1% SDS, 0.1M NaHCO3).
- Reverse Crosslinks & Purification: Incubate eluate at 65°C overnight with high salt to reverse crosslinks. Treat with RNase A and Proteinase K. Purify DNA using SPRI beads.
- Library Preparation & Sequencing: Prepare sequencing library from the purified DNA using a kit compatible with low-input samples. Sequence on an Illumina platform (≥20 million non-duplicate reads recommended).
Protocol 2: Native ChIP-seq for Histone Modifications
Principle: Uses micrococcal nuclease (MNase) digestion without crosslinking, ideal for stable epigenetic marks.
- Nuclei Isolation: Lyse cells in a gentle, non-ionic detergent buffer to isolate intact nuclei.
- MNase Digestion: Digest chromatin with MNase to yield primarily mononucleosomes. Quench with EGTA. Optimization Tip: Titrate MNase concentration to achieve >70% mononucleosomes.
- Chromatin Release & Immunoprecipitation: Release digested chromatin by nuclear lysis. Centrifuge to remove debris. Incubate soluble chromatin supernatant with 1-2 µg of anti-histone modification antibody overnight.
- Capture & Washes: Capture with Protein A/G magnetic beads. Wash with buffers of increasing stringency.
- DNA Elution & Purification: Elute DNA from beads. Purify using SPRI beads.
- Library Preparation & Sequencing: Construct libraries from the nucleosomal DNA. Sequence on an Illumina platform (≥10 million non-duplicate reads often sufficient for broad marks).
Visualizations
Diagram 1: Core ChIP-seq workflow
Diagram 2: TF binding and histone modification interplay
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for ChIP-seq
| Reagent/Material | Function & Application Note |
|---|---|
| High-Quality, Validated Antibodies | Specificity is paramount. Use ChIP-seq grade antibodies with published validation (e.g., ENCODE citations). Monoclonal preferred for TFs. |
| Magnetic Protein A/G Beads | For efficient capture of antibody-chromatin complexes. Offer low background and ease of handling over agarose beads. |
| Formaldehyde (37%) | For crosslinking protein-DNA and protein-protein interactions. Fresh aliquots are recommended. |
| Micrococcal Nuclease (MNase) | For native ChIP (nChIP) to digest linker DNA between nucleosomes. Requires careful titration. |
| SPRI (Solid Phase Reversible Immobilization) Beads | For consistent size selection and purification of DNA after elution and reverse crosslinking. |
| Low-Input Library Prep Kit | Essential for constructing sequencing libraries from often nanogram-scale ChIP DNA. |
| Cell Line/Tissue-Specific Lysis Buffers | Buffer composition (salt, detergent) must be optimized for the starting material to ensure clean nuclei isolation. |
| Protease/Phosphatase Inhibitor Cocktails | Critical to prevent degradation/modification of epitopes, especially for labile TFs or modifications. |
Application Notes
These components form the core of the Chromatin Immunoprecipitation (ChIP) process, a critical upstream step for ChIP-seq in epigenomics research. The quality and optimization of each directly determine the specificity, resolution, and signal-to-noise ratio of the final sequencing data, impacting downstream analyses of protein-DNA interactions, histone modifications, and transcription factor binding.
Antibodies: The primary determinant of specificity. A ChIP-grade antibody must have high affinity and specificity for the target epitope in its native, crosslinked chromatin context. Non-specific antibodies lead to high background and false-positive peaks.
Crosslinking: Typically using formaldehyde, this step creates covalent bonds between proteins and DNA, as well as between proximal proteins, "freezing" in vivo interactions. Under-crosslinking yields poor recovery; over-crosslinking creates a chromatin mesh resistant to sonication and masks epitopes.
Sonication: The method for fragmenting crosslinked chromatin to an optimal size (200–500 bp). This step determines the genomic resolution of the assay. Oversonication can damage epitopes and DNA, while undersonication reduces resolution and efficiency of IP.
Beads: Magnetic or agarose beads coated with Protein A, Protein G, or a recombinant fusion (e.g., Protein A/G) are used to capture antibody-target complexes. Bead choice depends on antibody species/isotype and requires optimization for binding capacity and minimal non-specific DNA retention.
Protocols
Protocol 1: Crosslinking & Chromatin Preparation for Cultured Cells
Materials: Formaldehyde (37%), Glycine (2.5 M), PBS, Lysis Buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5% NP-40, 0.25% Triton X-100), Shearing Buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.1% SDS).
Method:
- For adherent cells, add 1% final concentration of formaldehyde directly to culture media. Incubate 10 min at room temperature (RT) with gentle shaking.
- Quench crosslinking by adding glycine to a 125 mM final concentration. Incubate 5 min at RT.
- Wash cells twice with ice-cold PBS. Scrape and pellet cells (5 min, 500 x g, 4°C).
- Resuspend pellet in 1 mL Lysis Buffer. Incubate 10 min on a rotator at 4°C. Centrifuge (5 min, 1350 x g, 4°C). Discard supernatant.
- Resuspend pellet in 1 mL Shearing Buffer. Proceed immediately to sonication.
Protocol 2: Sonication for Chromatin Shearing (Covaris S220 Focused-ultrasonicator)
Materials: Covaris microTUBES (130 μL), Sheared chromatin, SPRIselect beads (Beckman Coulter).
Method:
- Transfer chromatin in Shearing Buffer to a Covaris microTUBE. Avoid bubbles.
- Use the following validated settings to achieve 200-500 bp fragments:
- Sonicate samples. Centrifuge tubes briefly to collect sample.
- Take a 20 μL aliquot for fragment analysis. To the remainder, add 1.1x volume SPRIselect beads, incubate, and purify on a magnet to remove debris and concentrate. Elute in 100 μL TE buffer.
Protocol 3: Immunoprecipitation with Magnetic Beads
Materials: Magnetic beads (Dynabeads Protein G), ChIP Blocking/Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl pH 8.0), Low Salt Wash Buffer (as above but 50 mM NaCl), High Salt Wash Buffer (as above but 500 mM NaCl), LiCl Wash Buffer (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH 8.0), Elution Buffer (1% SDS, 100 mM NaHCO3).
Method:
- Pre-clear: Add 20 μL of washed magnetic beads to 100 μL of sonicated chromatin. Rotate for 1 hr at 4°C. Capture beads on magnet, transfer supernatant to a new tube.
- Antibody Binding: Add recommended amount of antibody (typically 1–5 μg) to pre-cleared chromatin. Incubate 4–6 hrs at 4°C on a rotator.
- Bead Capture: Add 30 μL of pre-washed magnetic beads. Incubate 2 hrs at 4°C on a rotator.
- Washes: Capture beads and wash sequentially for 5 min each on rotator at 4°C with:
- 1 mL Low Salt Wash Buffer
- 1 mL High Salt Wash Buffer
- 1 mL LiCl Wash Buffer
- 2x with 1 mL TE buffer.
- Elution: Resuspend beads in 150 μL Elution Buffer. Incubate 30 min at 65°C with shaking (1000 rpm). Capture beads and transfer eluate (containing immunoprecipitated DNA-protein complexes) to a new tube.
Table 1: Quantitative Parameters for Key ChIP-seq Components
| Component | Optimal Parameter/Range | Impact of Deviation |
|---|---|---|
| Crosslinking (Formaldehyde) | 1% for 10 min (cell culture) | Short/Weak: Loss of transient interactions. Long/Strong: Reduced antibody access, poor sonication. |
| Sonication Fragment Size | 200–500 bp (avg. 300 bp) | Large (>700 bp): Poor genomic resolution. Small (<150 bp): DNA damage, loss of epitopes. |
| Antibody Amount | 1–5 μg per 10^6 cells | Low: Poor yield. High: Increased non-specific binding. |
| Magnetic Beads | 20–50 μL slurry per IP | Low: Incomplete capture. High: Increased non-specific background. |
| IP Wash Stringency | High Salt (500 mM NaCl) | Low Salt: High background. Excessive Salt: Disruption of specific interactions. |
Visualizations
ChIP-seq Experimental Workflow from Cells to Library
Core Immunoprecipitation Complex Assembly
The Scientist's Toolkit: Key Reagent Solutions
| Reagent/Material | Primary Function | Key Consideration for ChIP-seq |
|---|---|---|
| Formaldehyde (37%) | Reversible protein-protein and protein-DNA crosslinking. | Must be fresh; overuse leads to over-crosslinking. Quenching with glycine is critical. |
| ChIP-Validated Antibody | Binds specifically to the target antigen in fixed chromatin. | Must be validated for ChIP; check for citations, datasheets. The single largest source of failure. |
| Magnetic Beads (Protein A/G) | Solid-phase support to capture antibody-antigen complexes. | Protein A vs. G vs. A/G depends on antibody species/isotype. Low non-specific binding beads are essential. |
| Covaris Focused-Ultrasonicator | Shears crosslinked chromatin to precise, tunable fragment sizes. | Preferred over bath sonication for reproducibility and size targeting. Requires specific tubes and chillers. |
| SPRIselect Beads | Size-selective purification of DNA fragments; used post-sonication and post-IP. | Removes small fragments and contaminants. Ratios are critical for size selection. |
| Protease Inhibitor Cocktail | Prevents proteolytic degradation of proteins/chromatin during preparation. | Must be added fresh to all buffers prior to cell lysis and chromatin preparation. |
| RNAse A & Proteinase K | Enzymatic removal of RNA and proteins during DNA purification post-IP. | Essential for clean DNA recovery prior to library prep. |
| Dynabeads MyOne Streptavidin | Used in indexed ChIP methods (e.g., CUT&Tag, Low Cell # ChIP). | For capturing biotinylated DNA or nucleosome complexes. |
Within the context of a broader thesis on ChIP-seq protocol for epigenomics research, interpreting the biological meaning of a called "peak" is the critical final step. A peak in a ChIP-seq profile represents a genomic region enriched with sequenced DNA fragments from a Chromatin Immunoprecipitation (ChIP) experiment. This enrichment signifies the binding site of the protein of interest (e.g., transcription factor, histone modification) or the genomic locus associated with the chromatin feature being studied. However, a peak is not a direct molecular photograph; it is a statistical inference drawn from fragment pileup, requiring careful biological and technical interpretation.
The Multifaceted Meaning of a Peak
A peak's representation depends on the target of the antibody used.
Table 1: Interpretation of ChIP-seq Peaks Based on Target
| ChIP Target Type | What the Peak Primarily Represents | Typical Peak Shape | Key Considerations |
|---|---|---|---|
| Transcription Factor (TF) | Direct, sequence-specific DNA binding site of the protein. | Sharp, narrow (often 50-500 bp). | Requires high-quality antibody. Peaks often occur in promoter/enhancer regions. |
| Histone Modification (e.g., H3K27ac) | Genomic region marked by that epigenetic modification. | Broader regions (500-5000 bp). | Enrichment reflects density of nucleosomes carrying the mark. Represents active/repressive regulatory elements. |
| Histone Variant (e.g., H2A.Z) | Region enriched with nucleosomes containing that variant. | Broad. | Indicates dynamic or stable chromatin states. |
| Chromatin Regulator (e.g., Polycomb) | Binding site of the complex, often overlapping broad domains. | Can be mixed (sharp & broad). | May indicate recruitment sites or broader regulatory domains. |
| RNA Polymerase II | Transcriptionally active gene bodies and promoters. | Sharp peak at TSS, broad enrichment across gene. | Peak shape and location indicate initiation, pausing, or elongation. |
Key Experimental Protocols for Validation and Interpretation
Protocol: Verification of ChIP-seq Peak Specificity via Motif Analysis
Purpose: To determine if peaks from a TF ChIP-seq contain the known DNA binding motif, supporting direct binding. Materials: FASTA file of peak genomic sequences, motif discovery software (e.g., MEME-ChIP, HOMER). Procedure:
- Extract Sequences: Use
bedtools getfastato extract genomic sequences (e.g., ±100 bp from peak summit). - De Novo Motif Discovery: Input sequences into HOMER:
findMotifsGenome.pl peaks.bed <genome> output_dir -size 200. - Known Motif Enrichment: HOMER compares peaks to background genomic sequences for known motif enrichment.
- Interpretation: A statistically significant (p<1e-10) enrichment of the expected motif validates the experiment's specificity.
Protocol: Functional Validation of Candidate Regions by qPCR
Purpose: To independently confirm enrichment at specific peak loci. Materials: Original ChIP and Input DNA samples, qPCR reagents, primers designed for peak and negative control regions. Procedure:
- Primer Design: Design amplicons (80-150 bp) targeting:
- Peak Region: Center on the peak summit.
- Negative Control Region: A genomic locus without peaks (check via IGV).
- qPCR Setup: Perform SYBR Green qPCR in triplicate on ChIP and Input DNA for each primer set.
- Analysis: Calculate %Input for each region:
%Input = 2^(Ct[Input] - Ct[ChIP]) * Dilution Factor * 100. - Validation: Enrichment (%Input) at the peak region should be significantly higher (often >5-10x) than at the negative control region.
Protocol: Integrative Analysis with ATAC-seq or RNA-seq
Purpose: To interpret peaks in a functional genomic context (chromatin accessibility, gene expression). Materials: Processed ChIP-seq peak calls, ATAC-seq/RNA-seq data from the same/similar cell type. Procedure:
- Data Overlap: Use
bedtools intersectto find peaks overlapping ATAC-seq open chromatin peaks or gene promoters/TSS. - Correlation: For RNA-seq, associate TF peaks near gene TSS with expression changes upon TF perturbation.
- Visualization: Use IGV to co-visualize ChIP-seq, ATAC-seq, and RNA-seq (e.g., coverage tracks) at specific loci.
- Interpretation: A TF peak in an accessible region (ATAC-seq peak) near an upregulated gene suggests a functional binding event.
Title: Workflow for Interpreting ChIP-seq Peaks
Title: Peak Shape Reflects Underlying Biology
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for ChIP-seq Interpretation
| Item | Function & Rationale |
|---|---|
| High-Specificity ChIP-Validated Antibody | The cornerstone of the experiment. Must be validated for ChIP application to ensure peaks represent true target binding, not artifact. |
| Crosslinking Reagent (e.g., Formaldehyde) | Preserves transient protein-DNA interactions in vivo. Optimization of crosslinking time is critical for TFs vs. histones. |
| Chromatin Shearing Kit (Enzymatic or Sonicator) | Generates optimal fragment size (200-700 bp). Incomplete shearing reduces resolution; over-shearing destroys epitopes. |
| Magnetic Protein A/G Beads | Efficient capture of antibody-bound complexes. Reduce background vs. agarose beads. |
| Library Prep Kit for Low Input DNA | Post-ChIP DNA is scarce (<50 ng). Kits optimized for low-input improve library complexity and sequencing quality. |
| Peak Calling Software (e.g., MACS2) | Statistically identifies enriched regions vs. background (input control). Choice of parameters (q-value, shift) affects peak calls. |
| Genome Browser (e.g., IGV) | Essential for visual inspection of raw read pileup, peak shape, and integration with other genomic tracks. |
| Motif Analysis Suite (e.g., HOMER) | Identifies enriched DNA sequence motifs within peaks, confirming expected binding specificity. |
Step-by-Step ChIP-seq Protocol: From Cell Culture to Sequencing Library
The reproducibility and biological relevance of any Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) experiment are fundamentally determined in its initial phase. This phase establishes the foundation for robust epigenomic profiling by defining the requisite biological material, incorporating necessary experimental controls, and standardizing sample handling. Within the broader thesis on optimizing ChIP-seq for epigenomics research, this stage addresses the critical pre-analytical variables that can confound data interpretation, such as input DNA quality, antibody specificity, and cell state heterogeneity. Proper execution of Phase 1 is paramount for generating high-signal, low-noise datasets essential for drug discovery and mechanistic biology.
Quantitative Guidelines for Sample Preparation
Cell Number Requirements by Target & Species
The minimum number of cells required for a successful ChIP-seq experiment varies significantly based on the chromatin target's abundance and the model system. Current guidelines (updated 2023-2024) are summarized below.
Table 1: Recommended Cell Numbers for ChIP-seq
| Chromatin Target | Human/Mouse Cells | Drosophila / C. elegans Cells | Plant Cells (e.g., Arabidopsis) | Notes |
|---|---|---|---|---|
| Histone Modifications (H3K4me3, H3K27ac) | 50,000 - 200,000 | 10,000 - 50,000 | 100,000 - 500,000 | High-abundance marks; lower cell numbers feasible with optimized protocols. |
| Broad Histone Marks (H3K27me3, H3K9me3) | 100,000 - 500,000 | 20,000 - 100,000 | 200,000 - 1,000,000 | Wider genomic distribution requires more material for coverage. |
| Transcription Factors | 500,000 - 5,000,000 | 100,000 - 1,000,000 | 1,000,000 - 10,000,000 | Low abundance and transient binding necessitate high input. |
| RNA Polymerase II | 100,000 - 1,000,000 | 50,000 - 200,000 | 500,000 - 2,000,000 | Abundance depends on transcriptional activity of cells. |
| Archival FFPE Tissue | 1-3 tissue sections (5-10 μm thick) | N/A | N/A | Cell yield is highly variable; requires rigorous crosslink reversal and DNA repair. |
Control Experiment Specifications
A well-designed control strategy is non-negotiable for distinguishing specific enrichment from background.
Table 2: Essential Controls for ChIP-seq Experimental Design
| Control Type | Purpose | Recommended Specification | Protocol Reference |
|---|---|---|---|
| Input DNA | Controls for chromatin accessibility, sequencing bias, and genomic DNA contamination. | Use 1-10% of the volume/mass of chromatin used per IP. Must be processed alongside IP samples through crosslink reversal & purification. | See Protocol 3.1 |
| IgG (or pre-immune) | Negative control for non-specific antibody binding. | Use species-matched IgG, same concentration as specific antibody. Critical for identifying false-positive peaks. | See Protocol 3.2 |
| Positive Control Antibody | Validates overall ChIP procedure efficacy. | Use a well-characterized antibody (e.g., H3K4me3) on a reference cell line alongside experimental samples. | Standard IP protocol |
| Spike-in Chromatin | Normalizes for technical variation between samples (e.g., differential cell counts, IP efficiency). | Add defined amount of chromatin from a divergent species (e.g., Drosophila S2 cells to human cells) prior to IP. | See Protocol 3.3 |
| No Antibody Bead Control | Assesses background binding to beads/sepharose. | Incubate chromatin with beads only. | Standard IP protocol |
| Knockout/Degron Cell Line | Definitive control for antibody specificity. | Use genetically engineered cells lacking the target epitope. Gold standard but not always available. | N/A |
Detailed Protocols
Protocol 3.1: Input DNA Preparation
Objective: To generate a control sample representing the total population of sheared, crosslinked chromatin.
- After chromatin shearing and pre-clearing, remove an aliquot equivalent to 1-10% of the volume used for each IP reaction.
- Add 5M NaCl to a final concentration of 200mM and 1 μL of RNase A (10 mg/mL). Incubate at 65°C for 2 hours to reverse crosslinks.
- Add Proteinase K to a final concentration of 0.2 mg/mL. Incubate at 55°C for 30 minutes.
- Purify DNA using a PCR purification kit with elution in 30-50 μL of TE buffer or nuclease-free water.
- Quantify using a fluorometric assay (e.g., Qubit dsDNA HS Assay). Store at -20°C until library preparation.
Protocol 3.2: Negative Control IP with IgG
Objective: To quantify non-specific antibody and bead background.
- Prepare chromatin as for the specific IP.
- In a separate tube, use the same amount of chromatin, beads, and incubation buffers.
- Replace the specific antibody with an equivalent mass (typically 1-5 μg) of non-immune IgG from the same host species.
- Process the sample in parallel with the specific IP through all subsequent wash, elution, and crosslink reversal steps.
- Analyze the resulting DNA alongside the specific IP sample via qPCR at known positive and negative genomic loci before sequencing.
Protocol 3.3: Spike-in Chromatin Normalization (UsingDrosophilaS2 Chromatin)
Objective: To enable quantitative normalization between samples with varying starting material or IP efficiency.
- Spike-in Chromatin Preparation: Grow Drosophila melanogaster S2 cells to mid-log phase. Crosslink with 1% formaldehyde for 5 min. Quench, harvest, and lyse cells per standard protocol. Sonicate chromatin to 200-500 bp fragments. Aliquot and store at -80°C. Quantify DNA content.
- Spike-in Addition: Add a fixed, precise amount of S2 chromatin (e.g., 1% or 10% of the experimental chromatin by DNA mass) to each experimental human/mouse chromatin sample immediately before the IP step.
- Sequencing & Analysis: Sequence the IP material. During bioinformatics analysis, map reads to a combined reference genome (e.g., hg38 + dm6). Use the alignment statistics to the spike-in genome (dm6) to calculate normalization factors for the experimental genome (hg38) reads.
Visualization of Experimental Workflow & Controls
Title: Phase 1 ChIP-seq Workflow from Cells to Purified DNA
Title: Control Strategy for Robust ChIP-seq Data Interpretation
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for ChIP-seq Phase 1
| Item | Function & Rationale | Example Product/Type |
|---|---|---|
| Formaldehyde (37%) | Reversible crosslinker that fixes protein-DNA interactions. Critical for capturing transient binding events. | Ultra-pure, methanol-free grade. |
| Glycine (2.5M) | Quenches formaldehyde to stop crosslinking, preventing over-fixation and ensuring chromatin shearing efficiency. | Molecular biology grade. |
| Protease/Phosphatase Inhibitor Cocktails | Preserves the native state of chromatin and prevents post-lysis degradation or modification of target epitopes. | EDTA-free tablets or solutions. |
| Magnetic Protein A/G Beads | Solid support for antibody-antigen complex capture. Magnetic beads allow for rapid, clean wash steps. | Dynabeads, SureBeads. |
| Validated Primary Antibodies | Specific recognition of the chromatin target (histone mark, transcription factor, etc.). Validation for ChIP-seq is essential. | Cite-seq validated antibodies from major suppliers (e.g., Abcam, CST, Diagenode). |
| Non-immune IgG | Isotype control from the same host species as the primary antibody, required for the negative control IP. | Host species-matched (e.g., rabbit IgG). |
| Ultra-Sonicator | Instrument for chromatin fragmentation. Consistency and reproducibility of shearing are paramount for resolution and signal. | Focused ultrasonicator (e.g., Covaris M220) or Bioruptor. |
| DNA HS Assay Kit | Fluorometric quantification of low-concentration, sheared DNA. More accurate for ChIP DNA than absorbance (A260). | Qubit dsDNA HS Assay. |
| Spike-in Chromatin | Commercially prepared chromatin from a divergent species for cross-sample normalization. | Drosophila S2 or S. pombe chromatin kits. |
| PCR Purification Kit | For efficient purification and concentration of ChIP-enriched and Input DNA after crosslink reversal. | Column-based silica membrane kits. |
Within the context of optimizing Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) for epigenomics research, the choice of crosslinking strategy is fundamental. It dictates the balance between capturing transient protein-DNA interactions and maintaining chromatin accessibility for fragmentation and immunoprecipitation. This application note details the comparative use of standard formaldehyde (FA) versus dual crosslinkers (e.g., FA + DSG) for robust fixation, providing protocols and data to guide researchers and drug development professionals in stabilizing challenging epigenetic complexes.
Comparative Data Analysis
The efficacy of crosslinking strategies is quantified by metrics such as ChIP-seq library complexity, signal-to-noise ratio, and the recovery of specific genomic regions.
Table 1: Quantitative Comparison of Crosslinking Strategies for ChIP-seq
| Metric | Formaldehyde (FA) Alone | FA + Disuccinimidyl Glutarate (DSG) | Notes |
|---|---|---|---|
| Primary Target | Protein-DNA, RNA; short-range (2Å) | Protein-Protein (long-range, ~7.7Å) + Protein-DNA | DSG first stabilizes protein complexes, then FA fixes them to DNA. |
| Typical Efficiency for Histone Marks | High | Comparable to High | For stable, direct DNA binders. |
| Efficiency for Transcription Factors/Co-factors | Variable; can be low for indirect or transient binders | Significantly Enhanced | Dual crosslinking is critical for weak or chromatin-associated factors. |
| Chromatin Shearing Efficiency | Standard (requires optimization) | More Challenging (requires increased sonication) | Increased crosslinking density necessitates harsher fragmentation. |
| Background/Noise | Standard | Potentially Higher | Requires more stringent washes; can improve with optimized reversal. |
| Key Application | Routine histone mark ChIP-seq, strong DNA binders. | Challenging targets: non-DNA-binding co-regulators, chromatin remodelers, weak TFs. |
Table 2: Recommended Reversal Conditions
| Crosslinker | Reversal Condition | Incubation Time |
|---|---|---|
| Formaldehyde (FA) | 65°C with 200mM NaCl | 4-6 hours or overnight |
| FA + DSG | 65°C with 200mM NaCl | Overnight (12-16 hours) recommended |
Detailed Protocols
Protocol A: Standard Formaldehyde Crosslinking for Cell Cultures
Objective: To fix direct protein-DNA interactions for histone or strong TF ChIP-seq.
- Growth: Culture approximately 1x10^7 cells per IP to 70-80% confluence.
- Crosslinking: Add 37% formaldehyde directly to culture medium to a final concentration of 1%. Swirl gently.
- Incubate: Rock at room temperature (RT) for 10 minutes.
- Quenching: Add 2.5M glycine to a final concentration of 0.125M. Rock for 5 minutes at RT.
- Wash: Aspirate medium, wash cells twice with ice-cold PBS containing protease inhibitors.
- Harvest & Pellet: Scrape cells, pellet at 800 x g for 5 min at 4°C. Flash-freeze pellet or proceed to lysis.
Protocol B: Sequential DSG + Formaldehyde Dual Crosslinking
Objective: To stabilize both protein complexes and their DNA contacts for challenging epitopes.
- Preparation: Prepare a fresh 25mM stock of DSG (Disuccinimidyl Glutarate) in DMSO or DMF.
- Primary Crosslink (DSG): For adherent cells, replace medium with pre-warmed PBS containing 2mM DSG. Incubate for 45 minutes at RT with gentle rocking.
- Secondary Crosslink (FA): Without washing, add formaldehyde to the DSG/PBS solution to a final concentration of 1%. Rock for an additional 10 minutes at RT.
- Quenching & Wash: Quench with 0.125M glycine (final) for 5 min. Wash twice with ice-cold PBS.
- Harvest: Scrape and pellet cells. Process immediately or store at -80°C.
Note: For tissues, perform dicing and crosslinking in solution. Optimal DSG concentration (0.5-3mM) and time may require empirical testing.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Crosslinking Strategies
| Reagent/Material | Function | Example/Catalog Consideration |
|---|---|---|
| Formaldehyde (37%, Methanol-free) | Primary fixative; creates methylene bridges between amines. | Thermo Fisher Scientific, 28906 |
| Disuccinimidyl Glutarate (DSG) | Homobifunctional NHS ester; crosslinks primary amines between proteins. | Thermo Fisher Scientific, 20593 |
| Glycine | Quenches unreacted formaldehyde to stop crosslinking. | Standard molecular biology grade. |
| Protease Inhibitor Cocktail | Prevents protein degradation during cell processing. | EDTA-free (e.g., Roche cOmplete) |
| Sonicator (Covaris or tip-based) | Fragments crosslinked chromatin to desired size (200-600 bp). | Critical for shearing dual-crosslinked samples. |
| Micrococcal Nuclease (MNase) | Alternative for digesting chromatin prior to IP (native ChIP). | Used for some histone mark protocols. |
Visualizations
Title: Mechanism of Dual Crosslinking: DSG & Formaldehyde
Title: Experimental Workflow: FA vs. Dual Crosslinking ChIP-seq
Within the broader thesis, "A Standardized ChIP-seq Pipeline for Epigenomic Profiling in Drug Discovery," optimal chromatin fragmentation is a critical determinant of success. Sonication remains the predominant mechanical shearing method, balancing efficiency and practicality. Achieving the target 200-700 bp fragment range is paramount for two reasons: 1) Resolution: It ensures high mapping precision for transcription factor binding sites and histone modification peaks. 2) Immunoprecipitation Efficiency: Fragments that are too large (>1000 bp) reduce resolution and can lead to false-positive neighboring peaks, while excessively small fragments (<150 bp) may disrupt epitope integrity, reducing antibody capture. This application note details a systematic protocol for optimizing sonication parameters to achieve consistent fragment sizes.
Key Variables & Optimization Data
The primary variables influencing fragment size are sonication power (amplitude/duty cycle), total process time, and sample volume/viscosity. Optimization is instrument- and cell-type-specific. The following table summarizes quantitative findings from recent optimization experiments using a Covaris S220 focused-ultrasonicator and cultured HEK293 cells.
Table 1: Sonication Parameter Optimization for 200-700 bp Fragments (Covaris S220)
| Parameter | Tested Range | Optimal Value for HEK293 | Effect on Fragment Size |
|---|---|---|---|
| Peak Incident Power (W) | 105 - 175 | 140 | Higher power decreases average size. |
| Duty Factor (%) | 5 - 20 | 10 | Higher duty cycle increases shear energy, reducing size. |
| Cycles per Burst | 200 - 1000 | 200 | More cycles per burst increase energy, reducing size. |
| Treatment Time (s) | 45 - 180 | 120 | Longer time decreases average size; must be titrated. |
| Sample Volume (µL) | 50 - 200 | 130 | Consistent volume is critical for reproducible shear energy transfer. |
| Cell Count per Sample | 0.5M - 5M | 1-2 million | Higher chromatin concentration/viscosity requires more energy. |
| Temperature | 4-10°C | <6°C (maintained) | Prevents sample heating and chromatin degradation. |
Table 2: Expected Fragment Distribution Post-Optimization (Agarose Gel Analysis)
| Fragment Size Range (bp) | Percentage of Total | Suitability for ChIP-seq |
|---|---|---|
| < 150 bp | < 10% | Poor; may represent over-shearing/degradation. |
| 150 - 500 bp | > 60% | Ideal for high-resolution mapping. |
| 500 - 1000 bp | < 25% | Acceptable but may reduce mapping precision. |
| > 1000 bp | < 5% | Poor; requires extended sonication. |
Detailed Experimental Protocol
A. Pre-Sonication Chromatin Preparation
- Cell Fixation & Lysis: Harvest ~1x10⁶ cells per ChIP. Crosslink with 1% formaldehyde for 10 min at RT. Quench with 125 mM Glycine. Wash with cold PBS.
- Nuclear Lysis: Pellet cells. Resuspend in 1 mL Lysis Buffer 1 (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5% NP-40, 0.25% Triton X-100) and incubate 10 min on ice. Centrifuge. Resuspend pellet in 1 mL Lysis Buffer 2 (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) and incubate 10 min on ice. Centrifuge.
- Chromatin Resuspension: Resuspend nuclear pellet in 130 µL of Shearing Buffer (0.1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH 8.1). Transfer to a microTUBE (Covaris, cat# 520045).
- Pre-cooling: Place microTUBE in the Covaris filled with degassed, chilled water (4-6°C) for at least 5 minutes prior to sonication.
B. Titration Protocol for Sonication Optimization
- Prepare 6 identical chromatin samples from the same cell batch.
- Using the parameters in Table 1 as a starting point, vary only the treatment time across the samples (e.g., 45, 60, 90, 120, 150, 180 seconds).
- Perform sonication with the Covaris S220 set to: Peak Incident Power = 140W, Duty Factor = 10%, Cycles per Burst = 200.
- After sonication, centrifuge samples at 10,000g for 5 min at 4°C to pellet debris. Transfer supernatant to a new tube.
- Reverse crosslinks for 2 samples from each condition: Add 5 µL of 5M NaCl and 2 µL of 10 mg/mL RNase A, incubate at 65°C for 4 hours. Add Proteinase K, incubate at 45°C for 1 hour. Purify DNA via column purification.
- Analyze 20% of the purified DNA on a 1.5% agarose gel or a Bioanalyzer/Tapestation to generate a precise electrophoretogram. Identify the treatment time yielding the maximal concentration of fragments between 200-700 bp.
- Once optimal time is determined, fine-tune Duty Factor (± 2%) if the distribution is skewed too large or small.
C. Post-Sonication Processing for ChIP-seq
- Dilute sheared chromatin 10-fold in ChIP Dilution Buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH 8.1, 167 mM NaCl).
- Pre-clear with protein A/G beads for 1 hour at 4°C.
- Use 1-10 µg of chromatin per immunoprecipitation reaction, proceeding with the standard ChIP-seq protocol outlined in the broader thesis.
Visualization: Sonication Optimization Workflow
Title: Sonication Optimization & QC Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Chromatin Shearing Optimization
| Item | Function & Rationale | Example Product/Cat.# |
|---|---|---|
| Focused-Ultrasonicator | Delivers consistent, controllable acoustic energy for reproducible shear. Water bath cooling minimizes heating. | Covaris S220, E220 Evolution |
| microTUBE | Specific tube with precise geometry for optimal energy coupling and minimal sample loss. | Covaris microTUBE, AFA Fiber (520045) |
| High-Sensitivity DNA Assay | Accurate quantification and sizing of sheared, low-concentration chromatin DNA. | Agilent High Sensitivity DNA Kit (5067-4626) |
| SDS-Based Shearing Buffer | Contains mild detergent (SDS) to solubilize chromatin and facilitate uniform shearing. | 10 mM Tris, 1 mM EDTA, 0.1% SDS, pH 8.0 |
| Protein A/G Magnetic Beads | For pre-clearing and immunoprecipitation post-sonication; reduce non-specific background. | Pierce ChIP-Grade Protein A/G (26162) |
| Crosslinking Reagents | Reversible fixation of protein-DNA interactions. Formaldehyde is standard. | Ultrapure Formaldehyde (16% w/v), Methanol-free |
| Protease Inhibitor Cocktail | Prevents chromatin degradation by endogenous proteases during sample preparation. | cOmplete, EDTA-free (4693132001) |
| DNA Cleanup Columns | For post-reversal DNA purification prior to QC analysis. | SPRI/AMPure beads or silica-membrane columns |
Application Notes: In the Context of ChIP-seq for Epigenomics Immunoprecipitation (IP) is the cornerstone of the Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) workflow. The specificity of the antibody-target interaction and the stringency of the wash steps directly determine the signal-to-noise ratio and the validity of epigenetic data. Optimizing these parameters is critical for accurately mapping in vivo protein-DNA interactions, histone modifications, and transcription factor binding sites on a genome-wide scale.
1. Antibody Selection: The Primary Determinant of Specificity The choice of antibody is the most critical variable. For ChIP-seq, antibodies must recognize the target epitope in its native, crosslinked chromatin context.
Table 1: Antibody Characteristics for ChIP-seq
| Characteristic | Polyclonal | Monoclonal | Recombinant |
|---|---|---|---|
| Epitope Recognition | Multiple, good for modified residues (e.g., H3K27me3) | Single, high specificity for a single motif | Single, engineered for consistency |
| Specificity | Can vary between lots; higher risk of off-target binding | High and consistent between lots | Highest, engineered for minimal cross-reactivity |
| Affinity | Generally high due to multiple epitopes | Can be high, but is epitope-dependent | Engineered for optimal affinity |
| Recommended Use | Well-characterized histone modifications | Transcription factors, co-activators | Gold standard for reproducibility; any target |
| Validation Requirement | Essential (use knockout/knockdown controls) | Essential | Highly recommended |
Protocol 1.1: Antibody Validation for ChIP-qPCR
- Purpose: To confirm antibody specificity prior to scaling up for sequencing.
- Materials: Crosslinked chromatin (from target-positive and target-negative/isogenic knockout cells), Protein A/G magnetic beads, IP buffer, elution buffer.
- Method:
- Aliquot sheared chromatin (typically 2-5 µg per IP).
- Pre-clear chromatin with beads for 1 hour at 4°C.
- Incubate supernatant with 1-5 µg of test antibody and a matched isotype control IgG overnight at 4°C with rotation.
- Add beads and incubate for 2 hours.
- Wash beads with low-salt wash buffer (3x).
- Elute complexes, reverse crosslinks, and purify DNA.
- Perform qPCR on positive control genomic regions (known binding sites) and negative control regions (gene deserts, inactive loci). A valid antibody should show significant enrichment (>10-fold) over the IgG control at positive sites only.
2. Antibody Incubation: Optimizing Binding Dynamics Table 2: Incubation Parameter Optimization
| Parameter | Standard Condition | Optimization Guidance |
|---|---|---|
| Antibody Amount | 1-5 µg per 25-100 µg chromatin | Titrate (0.5 - 10 µg); balance between signal and background. |
| Incubation Time | Overnight (12-16 hours) at 4°C | Can reduce to 2-4 hours for high-affinity antibodies; longer may increase non-specific binding. |
| Temperature | Constant 4°C | Essential to preserve chromatin complexes and reduce degradation. |
| Buffer Volume & Agitation | 500 µL - 1 mL with end-over-end rotation | Ensure sufficient volume for mixing; avoid vortexing. |
3. Wash Stringency: Balancing Specificity and Yield Stringency is controlled by salt concentration, detergent type, and temperature during washes.
Table 3: Wash Buffer Stringency for ChIP-seq
| Buffer Type | Composition (Example) | Purpose & Stringency |
|---|---|---|
| Low-Salt Wash | 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 20 mM Tris-HCl pH 8.0 | Primary wash; removes non-specifically bound chromatin. Medium stringency. |
| High-Salt Wash | 500 mM NaCl, 0.1% SDS, 1% Triton X-100, 20 mM Tris-HCl pH 8.0 | Disrupts weak electrostatic interactions. High stringency. Use if background is high. |
| LiCl Wash | 250 mM LiCl, 1% NP-40, 1% Na-deoxycholate, 10 mM Tris-HCl pH 8.0 | Removes non-specific protein-protein interactions. High stringency. |
| TE Wash | 10 mM Tris-HCl, 1 mM EDTA pH 8.0 | Final rinse to remove detergents and salts before elution. Low stringency. |
Protocol 3.1: Stepwise Stringency Wash
- Purpose: To progressively remove non-specifically bound material while retaining true complexes.
- Method (Perform all steps at 4°C with tube rotation):
- After incubation, pellet beads and discard supernatant.
- Wash 1: Add 1 mL of Low-Salt Wash Buffer. Rotate for 5 minutes. Pellet beads, discard supernatant.
- Wash 2: Add 1 mL of High-Salt Wash Buffer. Rotate for 5 minutes. Pellet beads, discard supernatant.
- Wash 3: Add 1 mL of LiCl Wash Buffer. Rotate for 5 minutes. Pellet beads, discard supernatant.
- Wash 4: Add 1 mL of TE Buffer. Rotate for 2 minutes. Pellet beads, discard supernatant.
- Proceed to DNA elution.
Visualizations
Title: ChIP-seq IP Workflow Core Steps
Title: Increasing Wash Stringency to Isolate Specific Complexes
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Materials for ChIP-grade Immunoprecipitation
| Reagent / Solution | Function in the Protocol | Critical Consideration |
|---|---|---|
| Validated ChIP-seq Grade Antibody | Specifically binds the target protein or histone modification in fixed chromatin. | Primary driver of success. Seek citations from literature or vendor validation data. |
| Protein A/G Magnetic Beads | High-affinity capture of antibody-antigen complexes. Facilitate rapid wash steps. | Choose bead type (A, G, or A/G) based on the antibody species and subclass. |
| ChIP-Specific Lysis/Wash Buffers | Maintain complex integrity while removing non-specific interactions. | Buffer composition (salt, detergents) must be optimized for the target. |
| Protease & Phosphatase Inhibitors | Preserve the chromatin-bound protein complex during processing. | Must be added fresh to all buffers before use. |
| UltraPure BSA or Salmon Sperm DNA | Used as blocking agents to reduce non-specific bead binding. | Quality is vital to prevent introducing contaminants. |
| RNase A | Removes RNA that may co-purify with chromatin or cause viscosity. | Essential step before chromatin shearing for clean DNA isolation. |
| Glycogen or Carrier tRNA | Improves precipitation and recovery of low-concentration DNA during purification. | Critical for the final DNA elution step prior to library prep. |
The efficacy of a Chromatin Immunoprecipitation sequencing (ChIP-seq) experiment in epigenomics research is fundamentally dependent on the quality of the DNA library prepared for sequencing. Following immunoprecipitation, the protein-DNA complexes are crosslinked, and this reversal of crosslinks, coupled with the subsequent purification of DNA, is a critical bottleneck. Inefficient reverse crosslinking leads to poor DNA yield, while inadequate purification results in carryover of contaminants (proteins, salts, RNA, free nucleotides) that inhibit downstream enzymatic steps (e.g., adapter ligation, PCR). This application note details optimized protocols for these crucial steps, ensuring clean recovery of target sequences for high-fidelity NGS library construction in drug discovery and basic research.
Application Notes: Quantitative Comparison of Elution & Purification Methods
Table 1: Comparison of Reverse Crosslinking & Elution Conditions
| Condition | Temperature | Time | Additives | Avg. DNA Recovery (%) | PCR Inhibition (∆Ct) |
|---|---|---|---|---|---|
| Standard NaCl | 65°C | 4-6 hrs | 200 mM NaCl | 100% (Baseline) | 0 (Baseline) |
| High-Temp with SDS | 95°C | 10 min | 0.5% SDS | 95% | +0.8 |
| Proteinase K + High-Temp | 65°C → 95°C | 2 hrs → 15 min | Proteinase K (0.2 mg/mL) | 115% | -0.5 |
| RNase A Inclusion | 65°C → 95°C | 2 hrs → 15 min | Proteinase K + RNase A (0.1 mg/mL) | 118% | -1.2 |
Note: ∆Ct represents the change in qPCR threshold cycle compared to baseline, indicating inhibitor removal efficiency. Negative ∆Ct denotes improved amplification.
Table 2: Performance of DNA Purification Methods Post-Reverse Crosslinking
| Purification Method | Principle | Avg. Yield (%) | Fragment Size Retention | Residual Protein (ng/µL) | Suitability for Low Input |
|---|---|---|---|---|---|
| Phenol-Chloroform | Organic extraction | 70-80% | Excellent (>500 bp) | <1.0 | Moderate |
| Silica Spin Column | Binding in high salt | 60-75% | Bias >200 bp | <0.5 | Poor (High loss) |
| SPRI Beads (Size-Selective) | PEG/NaCl paramagnetic beads | 85-95% | Tunable (e.g., 100-500 bp) | <0.2 | Excellent |
| Ethanol Precipitation | Salting out | 50-70% | Good | 5.0-10.0 | Good |
Detailed Experimental Protocols
Protocol A: Optimized Reverse Crosslinking for ChIP Eluates
- Input: 50 µL of Protein G/A bead-antibody-target chromatin complex in elution buffer.
- Elution: Add 50 µL of Elution Buffer B (1% SDS, 0.1M NaHCO3). Vortex briefly.
- Incubate: Rotate at 65°C for 2 hours in a thermomixer (1000 rpm).
- Proteinase K Digestion: Add 2 µL of Proteinase K (20 mg/mL) for a final concentration of ~0.2 mg/mL. Mix thoroughly.
- Incubate: Rotate at 55°C for 30 minutes.
- Heat Inactivation/Reverse: Increase temperature to 95°C for 15 minutes.
- Cool: Briefly centrifuge tubes and place on ice.
- RNase Treatment (Optional): Add 1 µL of RNase A (10 mg/mL). Incubate at 37°C for 15 minutes.
Protocol B: SPRI Bead-based Cleanup & Size Selection
- Input: 100 µL of reverse-crosslinked DNA sample.
- Bead Preparation: Vortex SPRI beads thoroughly to achieve a homogeneous suspension.
- Binding: Add 90 µL (0.9X ratio) of SPRI beads to the sample. Mix by pipetting 10 times. Incubate at RT for 5 minutes. Note: A 0.9X ratio preferentially binds fragments >~150 bp.
- Capture: Place tube on a magnetic stand until supernatant is clear (~3 min). Discard supernatant.
- Wash: With tube on magnet, add 200 µL of freshly prepared 80% ethanol. Incubate 30 sec. Discard ethanol. Repeat wash once. Air-dry beads for 3-5 minutes.
- Elution: Remove tube from magnet. Elute DNA in 22 µL of 10 mM Tris-HCl, pH 8.0. Mix well. Incubate at RT for 2 minutes.
- Final Capture: Place tube on magnet. Transfer 20 µL of purified DNA to a fresh tube.
Visualizations
ChIP-seq DNA Recovery Workflow
SPRI Bead DNA Binding Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Reverse Crosslinking & Purification
| Item | Function & Critical Feature |
|---|---|
| Proteinase K (Recombinant, PCR-grade) | Digests histones and antibody proteins post-elution; essential for complete crosslink reversal. Must be RNase/DNase-free. |
| RNase A (DNase-free) | Removes co-precipitating RNA that can inflate QC measurements (Qubit/Bioanalyzer) and interfere with library prep. |
| SPRI (Solid Phase Reversible Immobilization) Beads | Polyethylene glycol (PEG)-coated magnetic beads for one-step cleanup and size selection. Ratio determines size cut-off. |
| Elution Buffer (1% SDS, 0.1M NaHCO3) | High-pH and detergent environment destabilizes protein-DNA interactions and initiates crosslink reversal. |
| Tris-EDTA (TE) Buffer, pH 8.0 | Low-salt, slightly basic elution buffer for final DNA resuspension; stabilizes DNA and is compatible with all NGS enzymes. |
| Magnetic Separation Stand | Enables efficient bead capture and supernatant removal during SPRI bead purification steps. |
| Thermonixer with Agitation | Provides consistent temperature and mixing during lengthy reverse crosslinking incubations, improving efficiency. |
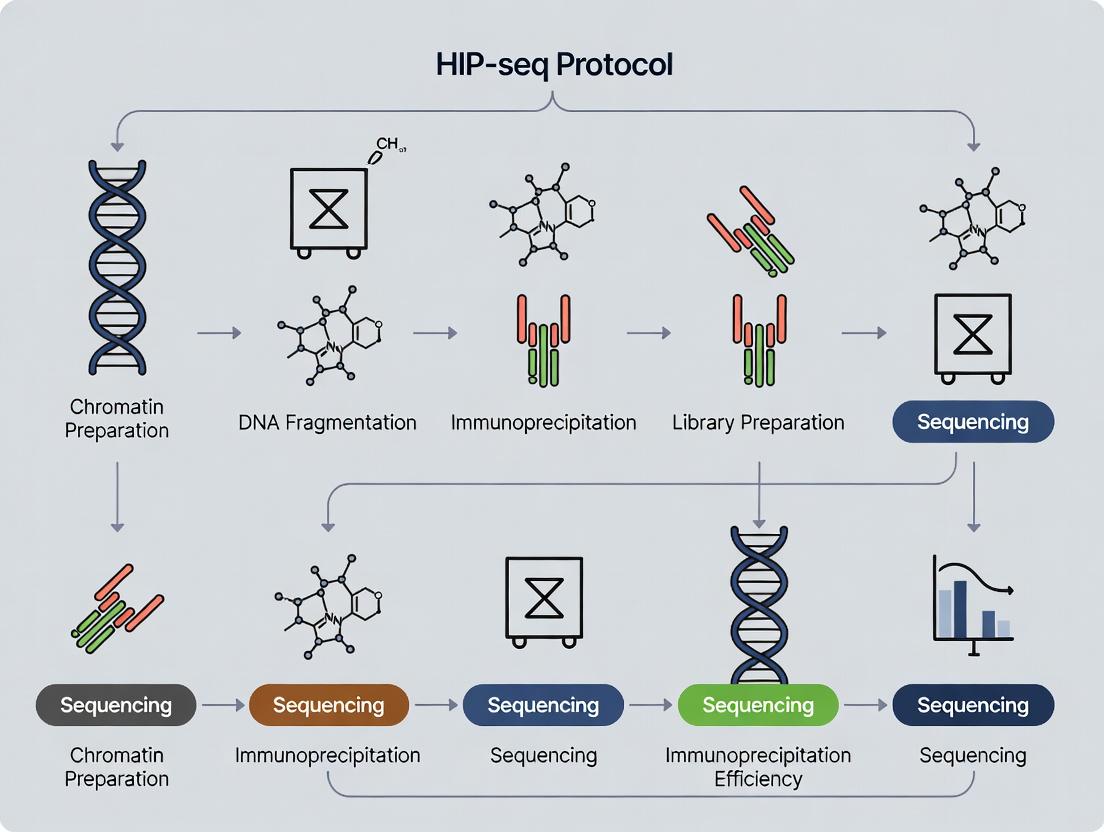
Within a ChIP-seq protocol for epigenomics research, the preparation of high-quality sequencing libraries is a critical determinant of data success. Following chromatin immunoprecipitation (ChIP), the purified DNA fragments must be converted into a format compatible with next-generation sequencing (NGS) platforms. This involves three core steps: size selection to isolate fragments of interest, adapter ligation to add platform-specific sequences, and amplification to generate sufficient material for sequencing. Optimal execution of these steps maximizes library complexity, minimizes bias, and ensures accurate mapping of protein-DNA interactions.
Size Selection Protocols
Size selection purifies DNA fragments within a desired range (typically 200–600 bp for standard ChIP-seq), removing very short fragments (e.g., primer dimers) and very long fragments. This improves sequencing efficiency and data resolution.
Protocol 1: Double-Sided SPRI Bead Cleanup
- Principle: Solid Phase Reversible Immobilization (SPRI) beads bind DNA in a size-dependent manner in the presence of polyethylene glycol (PEG) and salt.
- Detailed Methodology:
- First Bead Addition (Remove Large Fragments): To the purified ChIP DNA in a low-EDTA TE buffer, add SPRI beads at a ratio of 0.5x–0.7x sample volume. Mix thoroughly and incubate at room temperature for 5 minutes.
- First Supernatant Retention: Place tube on a magnet stand until supernatant clears. Transfer the supernatant (containing fragments smaller than the cutoff determined by the bead ratio) to a new tube. Discard beads.
- Second Bead Addition (Bind Target Fragments & Remove Small Fragments): To the supernatant, add SPRI beads at a ratio of 1.2x–1.5x the original sample volume. Mix and incubate for 5 minutes.
- Wash: Place on magnet, discard supernatant. With tube on magnet, wash beads twice with 200 µL of freshly prepared 80% ethanol.
- Elute: Air-dry beads for 2–5 minutes, then elute DNA in nuclease-free water or TE buffer.
Protocol 2: Agarose Gel Extraction
- Principle: DNA is separated by electrophoresis, and a gel slice containing the target size range is excised and purified.
- Detailed Methodology:
- Load the DNA sample alongside a DNA ladder on a 2% low-melt agarose gel.
- Run electrophoresis at low voltage (5–6 V/cm) until adequate separation is achieved.
- Visualize bands under low-energy UV light and excise the gel slice corresponding to the desired size range (e.g., 200–500 bp).
- Purify DNA from the gel slice using a commercial gel extraction kit, following manufacturer's instructions.
Table 1: Comparison of Size Selection Methods
| Method | Typical Size Range Recovery | Average Yield | Hands-on Time | Key Advantage | Key Disadvantage |
|---|---|---|---|---|---|
| Double-Sided SPRI Beads | Adjustable by bead ratio (e.g., 0.5x/1.2x yields ~200-600 bp) | High (>80%) | Low (~30 min) | Fast, scalable, automatable | Broader size distribution than gel |
| Agarose Gel Extraction | Precise (user-defined) | Moderate (50-70%) | High (~90 min) | High size precision, removes primer dimers effectively | Time-consuming, risk of UV damage |
| Pippin Prep System | Very precise (pre-set) | High (>80%) | Low (~20 min setup) | Automated, reproducible, high precision | Higher cost, requires specific cassettes |
Adapter Ligation Protocol
Adapters contain sequences required for cluster generation and sequencing on the NGS platform. Ligation attaches these adapters to both ends of the size-selected ChIP DNA.
Detailed Protocol for Ligation using Double-stranded DNA Adapters:
- Assemble Ligation Reaction: Combine components on ice in the following order:
- Size-selected DNA in water: X µL (typically 1–100 ng)
- Ligation Buffer (10X): 2.5 µL
- T4 DNA Ligase: 1.0 µL
- DNA Adapter Mix (diluted per manufacturer): 1.0 µL
- Nuclease-free water to a final volume of 25 µL.
- Incubate: Perform ligation at 20°C for 15 minutes for pre-annealed double-stranded adapters.
- Clean Up: Purify the ligated product using SPRI beads at a 1.0x ratio to remove excess free adapters. Elute in 20-25 µL of buffer.
Amplification Protocol
PCR amplification enriches for DNA fragments that have successfully ligated adapters on both ends and generates sufficient quantity for sequencing.
Detailed Protocol for Library Amplification:
- Assemble PCR Reaction:
- Ligated and purified DNA: 20 µL
- Universal PCR Primer Mix (10 µM each): 2.5 µL
- Index (Barcode) Primer (10 µM): 2.5 µL
- High-Fidelity PCR Master Mix (2X): 25 µL
- Total Volume: 50 µL.
- Perform Thermal Cycling:
- 98°C for 30 seconds (initial denaturation)
- Cycle 10-15 times:
- 98°C for 10 seconds (denaturation)
- 60°C for 30 seconds (annealing)
- 72°C for 30 seconds (extension)
- 72°C for 5 minutes (final extension)
- Hold at 4°C.
- Final Cleanup: Purify the amplified library using SPRI beads at a 0.9x ratio to remove PCR reagents and primers. Elute in 20-30 µL of low-EDTA TE buffer. Quantify by qPCR or bioanalyzer.
Table 2: Quantitative Metrics for Optimal ChIP-seq Library Prep
| Parameter | Optimal Range | Measurement Method | Impact on Sequencing Data |
|---|---|---|---|
| Input DNA Mass | 1–100 ng | Fluorometry (Qubit) | Lower input increases PCR duplicates, reduces complexity. |
| Final Library Yield | > 500 nM | qPCR (library-specific) | Ensures sufficient material for cluster generation. |
| Library Size Distribution | Peak: 250-350 bp | Bioanalyzer/TapeStation | Affects cluster density and mapping efficiency. |
| PCR Cycle Number | Minimum necessary (8-14) | - | High cycles increase duplicate rates and bias. |
| Adapter Dimer | < 5% of total signal | Bioanalyzer/TapeStation | Adapter dimers compete for sequencing reads. |
Visualizations
Title: ChIP-seq Library Prep Core Workflow
Title: Double-Sided SPRI Bead Size Selection
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for NGS Library Preparation in ChIP-seq
| Item | Function in ChIP-seq Library Prep | Example/Note |
|---|---|---|
| SPRI Beads | Magnetic beads for size selection and post-reaction cleanups. Enable buffer-based size fractionation. | AMPure XP, SPRIselect. Ratios are critical. |
| T4 DNA Ligase | Catalyzes the formation of phosphodiester bonds between DNA ends and compatible adapter overhangs. | Requires ATP. Often supplied with optimized buffer. |
| DNA Adapters (Indexed) | Short, double-stranded DNA oligos containing sequencing platform motifs and unique molecular barcodes. | Illumina TruSeq, IDT for Illumina. Must match platform. |
| High-Fidelity DNA Polymerase | PCR enzyme with low error rate and high processivity for limited-cycle amplification of libraries. | KAPA HiFi, NEBNext Q5. Minimizes PCR bias. |
| Size Selection Cassettes | Automated gel cassettes for precise, reproducible fragment isolation on systems like Pippin Prep. | Agarose gel alternative. Increases reproducibility. |
| Library Quantification Kit | qPCR-based assay using probes/primers specific to adapter sequences for accurate molarity. | KAPA Library Quant, NEBNext Library Quant. Critical for pooling. |
| Bioanalyzer/TapeStation | Microfluidics/capillary electrophoresis systems for assessing library size distribution and purity. | Agilent technologies. Detects adapter dimer contamination. |
In ChIP-seq for epigenomics, the choice between single-end (SE) and paired-end (PE) sequencing and the determination of appropriate sequencing depth are critical for accurately mapping protein-DNA interactions and histone modifications. This decision directly impacts data quality, resolution, and cost-efficiency within a drug development pipeline. SE reads are cost-effective for mapping transcription factor binding sites, while PE reads provide superior mapping accuracy in complex genomic regions and are often preferred for nucleosome positioning or histone mark studies. Sequencing depth must be calibrated to the biological question, with transcription factor studies requiring fewer reads than diffuse histone marks.
Quantitative Comparison: Sequencing Depth & Platform Metrics
Table 1: Recommended Sequencing Depth for ChIP-seq Targets
| ChIP-seq Target | Minimum Recommended Depth (SE) | Optimal Depth (PE) | Primary Rationale |
|---|---|---|---|
| Transcription Factors (e.g., p53) | 10-20 million reads | 20-30 million reads | Sharp, localized peaks; lower background. |
| Histone Marks (H3K4me3, H3K27ac) | 20-30 million reads | 30-50 million reads | Broad, enriched regions require more coverage. |
| Histone Marks (H3K36me3, H3K9me3) | 30-40 million reads | 40-60 million reads | Very broad domains necessitate high depth. |
| Input/Control | Matched to IP sample depth | Matched to IP sample depth | Essential for accurate peak calling and background subtraction. |
Table 2: Single-End vs. Paired-End Read Comparison for ChIP-seq
| Parameter | Single-End (SE) | Paired-End (PE) |
|---|---|---|
| Cost per Sample | Lower | ~1.5-2x SE cost |
| Mapping Accuracy in Repetitive Regions | Lower | Significantly Higher |
| Fragment Size Estimation | Indirect (modeled) | Direct from pair distance |
| Detection of Complex Events (e.g., rearrangements) | Limited | Possible |
| Ideal ChIP-seq Application | Transcription factor binding sites, QC assays | Histone marks, complex genomes, nucleosome positioning |
| Typical Read Length | 50-75 bp | 50-150 bp (each end) |
Detailed Protocols
Protocol 3.1: Determining Sequencing Depth for a Transcription Factor ChIP-seq Experiment
Objective: To establish the minimum sequencing depth required for robust peak calling of a transcription factor in a mammalian cell line.
Materials: See "The Scientist's Toolkit" below. Procedure:
- Pilot Experiment: Perform ChIP for the target transcription factor and prepare a sequencing library following standard cross-linking, sonication, immunoprecipitation, and library construction steps.
- Sequencing: Sequence the library to a moderate depth (e.g., 15 million SE reads).
- Bioinformatic Analysis: a. Align reads to the reference genome (e.g., using Bowtie2 or BWA). b. Call peaks using a tool like MACS2 with a matched input control. c. Generate a saturation analysis plot: - Randomly subsample sequencing reads (e.g., 10%, 20%, ...100%). - Call peaks on each subset. - Plot the number of identified peaks versus sequencing depth.
- Depth Determination: Identify the point where the curve plateaus. The depth just before this plateau is the minimum sufficient depth. For critical applications, add a 20-30% safety margin.
Protocol 3.2: Library Preparation for Paired-End ChIP-seq on a Diffuse Histone Mark
Objective: To generate a high-quality, strand-specific PE library for H3K36me3 ChIP-seq.
Procedure:
- Chromatin Shearing & IP: Perform ChIP as standard. Use Covaris sonication to shear cross-linked chromatin to a target size range of 150-300 bp. Immunoprecipitate with anti-H3K36me3 antibody.
- End Repair & A-tailing: Purify immunoprecipitated DNA. Perform end-repair to generate blunt-ended fragments, followed by addition of an 'A' base to the 3' ends using a polymerase.
- Adapter Ligation: Ligate indexed, strand-specific Y-shaped adapters to the A-tailed fragments. Purify to remove excess adapters.
- Size Selection: Perform double-sided size selection (e.g., using SPRI beads) to isolate fragments in the 200-400 bp range (including adapters). This is critical for PE sequencing.
- PCR Enrichment: Amplify the library with 10-15 cycles of PCR using primers complementary to the adapter sequences. Use a polymerase with high fidelity.
- Quality Control: Quantify the library by qPCR (for accurate molarity) and analyze fragment size distribution on a Bioanalyzer or TapeStation.
- Sequencing: Pool libraries and sequence on an Illumina platform with a 2x75 bp or 2x150 bp PE run. Aim for 50 million read pairs per sample as a starting point.
Visualization: Experimental Workflow & Decision Pathway
Title: ChIP-seq Sequencing Strategy Decision Pathway
Title: Core ChIP-seq Experimental Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for ChIP-seq Experiments
| Item | Function & Application | Example Vendor/Product |
|---|---|---|
| Anti-H3K27ac Antibody | Immunoprecipitation of specific histone modification for active enhancer profiling. | Abcam (ab4729), Cell Signaling Technology (8173S) |
| Protein A/G Magnetic Beads | Efficient capture of antibody-bound chromatin complexes; enables automation. | Thermo Fisher Scientific (10002D, 10004D) |
| Covaris Sonication System | Reproducible, controlled acoustic shearing of cross-linked chromatin to desired size. | Covaris (M220 Focused-ultrasonicator) |
| SPRIselect Beads | Solid-phase reversible immobilization for DNA purification, size selection, and cleanup. | Beckman Coulter (B23318) |
| Strand-Specific Sequencing Kit | Library preparation with unique molecular identifiers for accurate PE sequencing. | Illumina (TruSeq ChIP Library Prep Kit) |
| High-Fidelity PCR Polymerase | Accurate amplification of library fragments with minimal bias. | NEB (Q5 Hot Start), KAPA Biosystems (KAPA HiFi) |
| Bioanalyzer/TapeStation | Microfluidic analysis for precise quantification and size distribution of libraries. | Agilent (2100 Bioanalyzer) |
| Peak Calling Software | Computational identification of enriched genomic regions from aligned reads. | MACS2, HOMER, SPP |
Solving ChIP-seq Challenges: Troubleshooting Low Yield, High Background, and Artifacts
This application note, framed within our broader thesis on optimizing ChIP-seq for epigenomics research, provides a systematic guide for troubleshooting the critical signal-to-noise ratio. Poor specificity, manifesting as high background or low enrichment, often stems from issues in three core areas: antibody quality, crosslinking efficiency, or chromatin shearing.
Diagnostic Workflow & Decision Logic
Diagram Title: ChIP-seq Signal-to-Noise Diagnostic Decision Tree
Quantitative Benchmarks for Diagnosis
Table 1: Key QC Metrics and Target Ranges for ChIP-seq Components
| Component | QC Method | Optimal Range / Expected Result | Indication of Problem |
|---|---|---|---|
| Chromatin Shearing | Fragment Analyzer / Bioanalyzer | Majority 100-500 bp, peak ~200-300 bp | Majority >500 bp (under-sheared) or <150 bp (over-sheared) |
| Crosslinking Efficiency | DNA yield post-reversal (Input sample) | 1-10% of total chromatin DNA | Yield <<1% (over-XL) or >>10% (under-XL) |
| Antibody Efficacy | ChIP-qPCR (Positive Control Locus) | Enrichment ≥10x over IgG/Negative Control | Enrichment <5x over control |
| Antibody Specificity | ChIP-qPCR (Negative Control Locus) | Enrichment ~1x (same as IgG) | Enrichment >3x at negative locus |
Detailed Diagnostic Protocols
Protocol 1: Chromatin Shearing Optimization (Sonication)
Objective: Achieve uniform chromatin fragmentation (100-500 bp). Materials: Fixed cells, SDS lysis buffer, micrococcal nuclease (optional for combined approach), Covaris microTUBES or Diagenode Bioruptor tubes, sonicator (Covaris S220 or Diagenode Bioruptor Pico), Proteinase K, heat block. Procedure:
- Prepare 1x10^6 fixed cells in 130 µL SDS lysis buffer.
- Sonicate using a pre-optimized program. Example (Covaris S220): Peak Incident Power: 75W, Duty Factor: 10%, Cycles per Burst: 200, Time: 7 minutes (adjust based on cell type).
- Reverse crosslinks for 5 µL of sheared chromatin (add 2 µL 5M NaCl, 1 µL Proteinase K (20 mg/mL), incubate at 65°C for 2 hrs).
- Purify DNA (SPRI beads) and analyze on Fragment Analyzer.
- Adjust: If over-sheared, reduce time or power. If under-sheared, increase time, add a brief MNase digestion step (2-5 units, 37°C, 5 min) prior to sonication.
Protocol 2: Crosslinking Titration Test
Objective: Determine optimal formaldehyde concentration and duration. Materials: Cell culture, 16% or 37% Formaldehyde (methanol-free), 2.5M Glycine, PBS. Procedure:
- Aliquot 1x10^6 cells per condition into separate tubes.
- Crosslink with varying formaldehyde concentrations (0.5%, 1%, 1.5%) for a fixed time (e.g., 10 min) OR vary time (5, 10, 15 min) at a fixed concentration (e.g., 1%).
- Quench with 125 mM glycine (final conc.) for 5 min.
- Proceed with standard lysis and shearing (Protocol 1).
- Post-shearing, take a 10 µL "Input" sample, reverse crosslinks, purify DNA, and quantify by Qubit.
- Optimal condition: Yields DNA within the 1-10% range (Table 1) and produces the best fragment size profile after shearing.
Protocol 3: Antibody Validation via ChIP-qPCR
Objective: Confirm antibody specificity and enrichment power. Materials: Sheared chromatin, target antibody, species-matched IgG, Protein A/G beads, ChIP elution buffer, qPCR reagents, primers for known positive and negative genomic loci. Procedure:
- Perform standard ChIP using 1-5 µg of sheared chromatin per IP with 1 µg of test antibody or IgG.
- Wash, elute, and reverse crosslinks for all IP and 1% Input samples.
- Purify DNA and resuspend in 30 µL TE.
- Perform qPCR in triplicate for each sample using primers for a Positive Control Locus (e.g., active promoter for H3K4me3, gene desert for H3K9me3) and a Negative Control Locus.
- Calculate % Input and Fold Enrichment over IgG.
- Validation: A valid antibody shows high-fold enrichment at the positive locus and near-background at the negative locus (see Table 1).
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for ChIP-seq Troubleshooting
| Item | Function | Example Product (Supplier) |
|---|---|---|
| Methanol-free Formaldehyde | Reversible protein-DNA crosslinking; methanol can interfere. | Thermo Fisher Scientific (28906) |
| Validated ChIP-grade Antibody | Target-specific immunoprecipitation; critical for specificity. | Cell Signaling Technology (CST), Abcam, Diagenode |
| Magnetic Protein A/G Beads | Efficient antibody capture; low non-specific binding. | Dynabeads (Thermo Fisher) |
| Covaris microTUBES | Consistent acoustic shearing for optimal fragment size. | Covaris (520045) |
| SPRI Size Selection Beads | Cleanup and size selection of DNA fragments post-ChIP. | AMPure XP (Beckman Coulter) |
| Fragment Analyzer Kit | High-sensitivity analysis of DNA fragment size distribution. | High Sensitivity NGS Fragment Kit (Agilent) |
| Control Primer Sets | qPCR validation at known positive/negative genomic regions. | EpiTect ChIP qPCR Primer Assays (Qiagen) |
| Universal ChIP-seq Spike-in | Normalization across samples; identifies technical artifacts. | Spike-in Antibody (E2A Anti-Drosophila antibody), SNAP-ChIP (Cell Signaling) |
Optimizing for Low-Abundance Targets or Limited Input Material (MicroChIP)
Application Notes
Micro-ChIP (µChIP) addresses the critical challenge of performing chromatin immunoprecipitation with scarce biological samples, such as rare cell populations, fine-needle biopsies, or sorted stem cells. Its development has been pivotal for advancing epigenomics research in contexts where material is the limiting factor. This protocol is framed within the broader thesis that robust, scalable, and sensitive ChIP-seq methodologies are foundational for generating high-quality epigenomic maps, which in turn drive discoveries in gene regulation, disease mechanisms, and therapeutic targeting.
Recent advancements, as per current literature, emphasize microfluidic platforms, novel library amplification strategies, and enhanced background reduction to push the boundaries of sensitivity. Successful µChIP requires meticulous optimization at every step—from cross-linking and chromatin shearing to immunoprecipitation and library construction—to maximize signal-to-noise ratios while conserving material.
Table 1: Comparison of Key Low-Input ChIP-seq Methodologies
| Method | Typical Input Range | Key Innovation | Primary Advantage | Reported Sensitivity (Post-IP DNA Yield) |
|---|---|---|---|---|
| Standard ChIP-seq | 0.5-10 million cells | N/A | Benchmark protocol | 10-100 ng |
| MicroChIP (µChIP) | 1,000 - 100,000 cells | Downscaled volumes, carrier materials | Adapts standard protocols | 0.1-5 ng |
| ULI-NChIP | 100 - 10,000 cells | Native ChIP, no crosslinking | High resolution for histones | 0.01-1 ng |
| TELP / ChIPmentation | 500 - 50,000 cells | In-tagmentation via Tn5 transposase | Faster, fewer steps | 0.05-2 ng |
| MOWChIP-seq | 100 - 10,000 cells | Microfluidics on a bead-packed chip | Automated, minimal handling | 0.02-0.5 ng |
| CUT&RUN / CUT&Tag | 100 - 100,000 cells | In situ cleavage by pA-Tn5 fusion | Exceptionally low background | Not applicable (direct tagmentation) |
Experimental Protocols
Protocol 1: MicroChIP for Histone Modifications (10,000 Cells)
Cell Preparation and Crosslinking:
- Pellet 10,000 cells. Resuspend in 45 µL PBS.
- Add 5 µL of 37% formaldehyde (final concentration 1%). Mix and incubate at room temperature for 8 minutes.
- Quench with 5 µL of 2.5M glycine (final concentration 125 mM). Incubate 5 minutes at RT.
- Pellet cells at 600 x g for 5 min at 4°C. Wash twice with 1 mL ice-cold PBS. Pellet can be frozen at -80°C.
Chromatin Shearing (Critical for Low Input):
- Lyse cells in 50 µL lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1) with protease inhibitors.
- Sonicate using a focused ultrasonicator with microTUBEs. Perform 4 cycles of 30 sec ON / 30 sec OFF at peak power. Keep samples at 4°C.
- Centrifuge at 20,000 x g for 10 min at 4°C. Transfer supernatant (sheared chromatin) to a new tube. Dilute 10x with IP Dilution Buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH 8.1, 167 mM NaCl).
- Take a 2 µL aliquot to check fragment size (target 150-500 bp) via a High Sensitivity DNA Bioanalyzer chip.
Immunoprecipitation and Wash:
- Pre-clear chromatin with 20 µL of protein A/G magnetic beads for 1 hour at 4°C.
- Incubate supernatant with 0.5-1 µg of target-specific antibody (e.g., anti-H3K4me3) overnight at 4°C with rotation.
- Add 25 µL of pre-blocked protein A/G magnetic beads and incubate for 2 hours.
- Wash beads sequentially with 200 µL of each wash buffer for 5 minutes on rotation:
- Low Salt Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.1, 150 mM NaCl)
- High Salt Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.1, 500 mM NaCl)
- LiCl Wash Buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH 8.1)
- Two washes with TE Buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0)
Elution and Clean-up:
- Elute chromatin from beads twice with 50 µL of Freshly Prepared Elution Buffer (1% SDS, 100 mM NaHCO3), incubating at 65°C for 15 minutes with shaking.
- Combine eluates (100 µL total). Add 4 µL of 5M NaCl and reverse crosslinks by incubating at 65°C overnight.
- Add 2 µL of 1M Tris-HCl (pH 6.5), 2 µL of 0.5M EDTA, and 1 µL of 20 mg/mL Proteinase K. Incubate at 55°C for 2 hours.
- Purify DNA using a silica-membrane column kit designed for recovery of low-abundance DNA (e.g., ChIP DNA clean & concentrator). Elute in 12 µL nuclease-free water.
Protocol 2: Low-Input Library Construction for ChIP-seq (ThruPLEX-like Method)
This protocol uses a tagmentation-based template switching approach for maximal efficiency from sub-nanogram inputs.
- End Repair & A-Tailing: To the 12 µL ChIP DNA, add 8 µL of a master mix containing DNA polymerase, dNTPs, and ATP. Incubate at 20°C for 30 min, then 65°C for 30 min.
- Adapter Ligation: Add 20 µL of ligation master mix containing stem-loop adapters with a blocking group. Incubate at 20°C for 15 min. Add a reagent to cleave the blocking group.
- PCR Amplification: Add 25 µL of PCR master mix containing barcoded primers and high-fidelity polymerase. Cycle as follows:
- 72°C for 3 min
- 98°C for 2 min
- Cycle 10-16x: 98°C for 15 sec, 60°C for 30 sec, 72°C for 30 sec
- 72°C for 1 min.
- Clean-up: Purify amplified libraries using SPRi beads at a 1:1 ratio. Elute in 20 µL TE buffer.
- Quality Control: Assess library size distribution on a High Sensitivity DNA Bioanalyzer chip and quantify via qPCR with a library quantification kit.
Visualizations
Workflow for MicroChIP and Sequencing
Strategies to Reduce Background in MicroChIP
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for MicroChIP
| Item | Function in MicroChIP | Key Consideration for Low Input |
|---|---|---|
| High-Sensitivity Sonication System (e.g., focused ultrasonicator with microTUBEs) | Efficient chromatin shearing to ideal fragment sizes (200-500 bp) with minimal sample loss. | Micro-volume containers are essential to prevent adsorption to walls and maximize shearing efficiency. |
| Validated High-Titer ChIP-Grade Antibody | Specific recognition of the low-abundance chromatin target (e.g., transcription factor, histone mark). | Affinity and specificity are paramount; high background from poor antibodies is catastrophic with limited material. |
| Magnetic Beads (Protein A/G) | Capture and wash of antibody-chromatin complexes. | Use pre-blocked beads with BSA and carrier DNA/RNA (e.g., yeast tRNA, salmon sperm DNA) to reduce non-specific binding. |
| Low-Input DNA Library Preparation Kit (e.g., ThruPLEX, SMART-ChIP, or tagmentation-based) | Amplification of picogram DNA to sequencing-ready libraries with minimal bias and duplication. | Kit selection is critical; must have high efficiency from sub-nanogram inputs and maintain complexity. |
| High-Sensitivity DNA Analysis Kits (e.g., Bioanalyzer HS DNA, TapeStation HS D1000) | QC of sheared chromatin and final library size distribution. | Standard agarose gels lack the sensitivity to visualize low-input ChIP DNA prior to library prep. |
| Silica-Membrane or SPRI Bead Clean-up Kits | Purification of DNA after elution and between library prep steps. | Optimize bead-to-sample ratios for small fragment recovery; avoid over-drying which reduces elution efficiency. |
| Carrier Substances (e.g., Glycogen, Yeast tRNA) | Co-precipitation agent to visually track and improve recovery during ethanol precipitation steps. | Use PCR-inert carriers if used prior to library amplification to avoid inhibition or contamination. |
Within the broader thesis on optimizing ChIP-seq protocols for epigenomics research, a paramount challenge is mitigating high background signals. Non-specific antibody binding and non-target protein-DNA interactions generate noise that obscures true epigenetic marks, compromising data integrity and biological interpretation. This application note details contemporary strategies and specificity controls essential for robust, publication-quality ChIP-seq.
Core Challenges & Quantitative Impact
High background in ChIP-seq manifests as elevated signal in negative control samples, reducing peak-to-background ratios and increasing false-positive rates. The table below summarizes common causes and their quantitative impact on data quality.
Table 1: Common Sources of High Background in ChIP-seq and Their Impact
| Source of Background | Typical Manifestation | Approximate Impact on Signal-to-Noise (Untreated vs. Addressed) |
|---|---|---|
| Non-specific Antibody Binding | High signal in IgG/isotype control | Can reduce SNR by 50-80% |
| Insufficient Chromatin Shearing | Large DNA fragments (>1000 bp) | Increases background reads by 2-5 fold |
| Inadequate Blocking | High signal in no-antibody control | Can increase false positives by 3-10x |
| Cross-linked Protein Aggregates | High signal in pre-clearing flow-through | Reduces mappable reads by 20-40% |
| Endogenous Biotin/Sticky Sites | Enrichment in negative genomic regions | Region-dependent; can cause >100 false peaks |
Research Reagent Solutions Toolkit
Table 2: Essential Reagents for Background Reduction
| Reagent / Material | Primary Function | Key Consideration |
|---|---|---|
| Protein A/G Magnetic Beads | Immunoprecipitation of antibody complexes | Pre-blocking with BSA/sheared salmon sperm DNA is critical. |
| Species-Matched IgG | Isotype control for specificity | Must match host species, subclass, and conjugation of primary antibody. |
| Sheared Salmon Sperm DNA / BSA | Blocking agent for beads & assay | Competes for non-specific DNA/protein binding sites. |
| Protease/Phosphatase Inhibitor Cocktails | Preserves complex integrity | Prevents degradation and aberrant protein-DNA interactions. |
| Recombinant Protein A/G | Pre-clearing agent | Removes antibodies that bind beads non-specifically. |
| Digitonin | Permeabilization agent (for nuclei) | Cleaner than NP-40 for native ChIP; reduces cytoplasmic contamination. |
| Glycogen or tRNA | Carrier for DNA precipitation | Inert, reduces loss of low-concentration DNA. |
| RNase A | Removes RNA | Prevents co-precipitation of RNA-bound proteins. |
| Triton X-100 / SDS | Detergents for lysis & washing | Optimal concentration is cell-type specific; affects background. |
Specificity Controls: Protocols & Implementation
Protocol 1: Isotype Control (IgG) ChIP
Objective: To measure non-specific antibody binding and background DNA precipitation.
- Parallel Sample Preparation: Split sheared, cross-linked chromatin into two equal aliquots (e.g., 10 µg each).
- Immunoprecipitation: To one aliquot, add the specific target antibody (e.g., anti-H3K27ac). To the other, add an equivalent amount (µg) of species- and subclass-matched non-immune IgG.
- Identical Processing: Subject both samples to identical subsequent steps: incubation with pre-blocked beads, washing, elution, reverse cross-linking, and DNA purification.
- Sequencing & Analysis: Sequence both libraries. The IgG control provides a background model. True peaks must be significantly enriched over the IgG profile (e.g., ≥5-fold enrichment, FDR < 1%).
Protocol 2: "No-Antibody" & Beads-Only Control
Objective: To assess background from non-specific chromatin sticking to beads or tubes.
- Sample Setup: Prepare a third aliquot identical to those in Protocol 1.
- Omission: Process this sample through the entire IP protocol but omit the primary antibody. Include the bead-blocking and washing steps.
- Analysis: The resulting DNA library quantifies chromatin that binds to magnetic beads or tube walls independently of an antibody. This should yield negligible DNA (e.g., <0.01% of input).
Protocol 3: Input DNA Reference
Objective: To control for chromatin accessibility and sequence bias in shearing/PCR.
- Reservation: Before IP, reserve 1-10% of the sheared, cross-linked chromatin.
- Processing: Reverse cross-link this "Input" sample, treat with RNase A and Proteinase K, and purify the DNA alongside the IP samples.
- Use: The Input is sequenced to map open chromatin regions and copy number variations. It is the critical denominator for peak-calling algorithms (e.g., MACS2).
Blocking Strategies: Detailed Methodologies
Strategy A: Pre-blocking of Magnetic Beads
Rationale: Beads (e.g., Protein A/G) have surface sites that bind biomolecules non-specifically.
- Wash beads 3x in ChIP IP Buffer (e.g., 20 mM Tris-HCl pH 8.0, 140 mM NaCl, 1% Triton X-100, 0.1% SDS).
- Resuspend beads in 1 mL IP Buffer containing 0.5 µg/µL BSA and 0.1 µg/µL sheared salmon sperm DNA.
- Rotate at 4°C for 2 hours.
- Wash beads 3x with 1 mL IP Buffer to remove excess block. Use immediately for IP.
Strategy B: Chromatin Pre-clearing
Rationale: Removes chromatin fragments that bind non-specifically to the bead matrix.
- After chromatin shearing and dilution in IP buffer, add 10-20 µL of pre-blocked beads (without antibody).
- Rotate the mixture for 1-2 hours at 4°C.
- Place tube on a magnet and carefully transfer the supernatant to a new tube. This pre-cleared chromatin is used for the subsequent IP steps.
Strategy C: Optimized Wash Buffer Series
Rationale: A stepwise increase in stringency removes loosely bound complexes. Protocol: Perform all washes cold (4°C) with rotation for 3-5 minutes.
- Low Salt Wash: 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS. (2x)
- High Salt Wash: 20 mM Tris-HCl pH 8.0, 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS. (1x)
- LiCl Wash: 10 mM Tris-HCl pH 8.0, 250 mM LiCl, 1 mM EDTA, 1% NP-40, 1% Sodium Deoxycholate. (1x)
- TE Wash: 10 mM Tris-HCl pH 8.0, 1 mM EDTA. (2x)
Data Interpretation: From Controls to Confident Peaks
Effective use of controls allows for rigorous bioinformatic thresholding. The table below outlines key quality metrics derived from control experiments.
Table 3: Quantitative Metrics for Assessing ChIP-seq Specificity
| Metric | Calculation | Optimal Range | Indication of Problem |
|---|---|---|---|
| FRiP (Fraction of Reads in Peaks) | (Reads in peaks) / (Total mapped reads) | 1-30% (target-dependent) | <1% suggests poor enrichment or high background. |
| Signal-to-Noise Ratio (SNR) | (Reads in target IP) / (Reads in IgG control) | ≥5 for strong marks (e.g., H3K4me3) | <3 indicates poor specificity. |
| Peak Shift Quality | Fragment length distribution from cross-correlation | Strong bimodal distribution | Single broad peak suggests poor shearing or background. |
| % of Blacklisted Regions | Peaks overlapping ENCODE blacklists (e.g., satellite repeats) | <1-2% | >5% indicates non-specific or artefactual binding. |
Visual Summaries
Title: ChIP-seq Specificity Control Workflow
Title: Specific vs. Non-specific Complex Wash Stringency
Integrating the described specificity controls (IgG, no-antibody, Input) with proactive blocking strategies (bead pre-blocking, chromatin pre-clearing, stringent washes) is non-negotiable for definitive epigenomics research. This systematic approach quantitatively minimizes background, transforming high-noise ChIP-seq data into a reliable map of protein-DNA interactions, thereby strengthening the foundational data of the overarching thesis.
Mitigating PCR Duplicates and Sequencing Biases in Library Amplification
Abstract Within a ChIP-seq workflow for epigenomics research, the final library amplification step is critical yet prone to introducing artifacts. PCR duplicates can inflate read counts, skewing quantitative analyses of histone modifications or transcription factor binding. Furthermore, sequence-dependent amplification biases can distort the true representation of genomic fragments. This application note details strategies and optimized protocols to minimize these artifacts, ensuring data integrity for downstream discovery and validation in drug target identification.
Understanding and Quantifying Artifacts
1.1. PCR Duplicates PCR duplicates are identical copies of an original DNA fragment, formed during library amplification. In ChIP-seq, they are identified by matching genomic coordinates and, crucially, unique molecular identifiers (UMIs). High duplicate rates (>50%) often indicate low input material or over-amplification, confounding peak calling and quantitative comparisons.
1.2. Sequence-Specific Bias GC content and secondary structure affect polymerase efficiency, leading to uneven coverage. This bias is particularly problematic in open chromatin regions or specific motif-dense areas, potentially creating false-positive or false-negative peaks.
Table 1: Impact and Identification of Amplification Artifacts
| Artifact Type | Primary Cause | Typical Rate in ChIP-seq | Downstream Impact | Detection Method |
|---|---|---|---|---|
| PCR Duplicates | Over-amplification, low input | 20-50% (input-dependent) | Inflated read counts, skewed quantification | UMI-based deduplication; coordinate-based marking (without UMIs) |
| GC Bias | Differential polymerase efficiency across GC% | Coverage variance up to 40% | False enrichment/depletion in GC-rich/poor regions | Pre-sequencing qPCR bias assays; post-sequencing coverage analysis |
| Adapter Dimer | Excessive cycles, inefficient cleanup | 5-15% of reads (if severe) | Loss of sequencing throughput, background noise | Bioanalyzer/TapeStation peak ~128bp |
Core Mitigation Strategies & Protocols
2.1. Strategy A: UMI Integration for Duplicate Identification Incorporating Unique Molecular Identifiers (UMIs) during adapter ligation allows precise identification of true biological molecules.
Protocol: UMI-Adapter Ligation for ChIP-seq Libraries Materials: Purified ChIP DNA, UMI-containing dual-indexed adapters, ligase, PCR reagents, size-selection beads.
- End Repair & A-tailing: Perform standard end-prep on ChIP DNA.
- UMI Adapter Ligation: Ligate UMI adapters. Use a 10-15x molar adapter excess to input DNA. Critical Step: Minimize PCR cycles post-ligation.
- Clean-up: Purify with 1.0x bead ratio to remove adapter dimers.
- Limited-Cycle Enrichment PCR: Amplify with polymerase suitable for complex genomic DNA. Use as few cycles as possible (typically 4-10). Determine optimal cycles via qPCR side reaction.
- Final Purification: Size-select for 200-700 bp fragments.
2.2. Strategy B: Bias-Reducing Polymerase and Buffer Systems Using engineered polymerases and optimized buffers minimizes GC bias.
Protocol: Optimization of Amplification for GC-Rich Regions Materials: Bias-reducing polymerase mix (e.g., KAPA HiFi, Q5), GC enhancer additives, magnetic beads.
- Setup Test Reactions: Aliquot identical library prep samples pre-amplification.
- Condition Testing: Set up parallel reactions:
- Condition 1: Standard polymerase.
- Condition 2: Bias-reducing polymerase.
- Condition 3: Bias-reducing polymerase + 1x GC enhancer.
- qPCR Monitoring: Use SYBR Green qPCR with a primer for a known, difficult-to-amplify GC-rich locus and a control locus. Calculate ΔCq.
- Cycle Determination: Amplify until just entering plateau phase. Use the condition with the smallest ΔCq between GC-rich and control loci for the full-scale reaction.
2.3. Strategy C: Linear Amplification Methods Linear amplification avoids the exponential duplication issue.
Protocol: In Vitro Transcription (IVT)-Based Amplification Materials: T7 promoter-containing adapter, T7 RNA polymerase, RNA-to-cDNA conversion kit.
- Adapter Ligation: Ligate ChIP DNA to a double-stranded adapter containing a T7 promoter sequence.
- Linear RNA Amplification: Perform IVT reaction to generate multiple RNA copies from each template.
- Reverse Transcription: Convert amplified RNA back to cDNA using random primers and reverse transcriptase.
- Second Strand Synthesis & Final Library Prep: Generate dsDNA and perform a limited-cycle (2-4) PCR to add sequencing indices.
Table 2: Comparative Evaluation of Mitigation Protocols
| Strategy | Key Reagent | Optimal Input | Estimated Duplicate Reduction | Bias Mitigation | Complexity/Cost |
|---|---|---|---|---|---|
| UMI + Limited PCR | UMI Adapters, High-Fidelity Polymerase | Moderate to High (5-50 ng) | 70-90% (via bioinformatic removal) | Moderate | Medium |
| Bias-Reducing Polymerase | Engineered Polymerase Mix | Any | 30-50% (by reducing required cycles) | High (GC bias) | Low |
| Linear Amplification (IVT) | T7 RNA Polymerase | Very Low (<1 ng) | >90% (minimal exponential PCR) | Moderate | High |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Mitigating Amplification Artifacts
| Reagent/Material | Function & Role in Artifact Mitigation | Example Product |
|---|---|---|
| UMI Dual-Indexed Adapters | Uniquely tags each original molecule pre-amplification for precise duplicate identification. | IDT for Illumina UDI adapters |
| High-Fidelity/ Bias-Reducing Polymerase | Engineered for even amplification across varying GC content, reducing coverage bias and allowing fewer cycles. | KAPA HiFi HotStart, NEB Q5 Ultra II |
| GC Enhancer Additive | Destabilizes secondary structure, improving polymerase processivity in high-GC regions. | Q5 GC Enhancer, KAPA GC Boost |
| Solid Phase Reversible Immobilization (SPRI) Beads | For precise size selection and clean-up, critical for removing adapter dimers that consume cycles. | Beckman Coulter AMPure XP |
| T7 Promoter Adapter & IVT Kit | Enables linear RNA amplification, drastically reducing PCR duplicate formation from low-input samples. | NEB Next Ultra II RNA Library Prep |
Workflow and Pathway Visualizations
Title: ChIP-seq Workflow with UMI Integration
Title: qPCR Assay for GC Bias Detection
Protocol Adaptations for Difficult Tissues, FFPE Samples, or Rare Cell Populations
Within the broader thesis on advancing ChIP-seq methodology for epigenomics research, a significant challenge lies in the robust analysis of suboptimal or limited starting materials. This application note details optimized protocols and analytical considerations for three critical scenarios: difficult-to-lyse tissues (e.g., fibrous, fatty), formaldehyde-fixed paraffin-embedded (FFPE) archives, and rare cell populations. These adaptations are essential for expanding epigenetic analysis to clinically relevant samples and rare disease models, thereby bridging the gap between foundational epigenomics and translational drug development.
Standard ChIP-seq protocols assume the availability of millions of fresh, homogeneous cells. However, biologically crucial questions often involve samples that deviate from this ideal—archival FFPE blocks, minute biopsies, or rare circulating tumor cells. The core thesis of this work posits that with targeted modifications to chromatin preparation, immunoprecipitation, and library construction, high-quality epigenetic data can be recovered from these challenging sources. Success hinges on understanding and mitigating the specific liabilities of each sample type.
Protocol for Difficult-to-Process Tissues
Fibrous (heart, muscle), fatty (adipose, brain), or sclerotic tissues resist standard lysis, leading to low chromatin yield and fragmentarity.
Detailed Methodology: Mechanical & Enzymatic Disruption
- Tissue Preparation: Snap-freeze tissue in liquid N₂. Pulverize using a cryogenic impact grinder (e.g., Covaris cryoPREP) to a fine powder.
- Dual Lysis: Suspend powder in 1 mL LB1 (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100) and homogenize with a Dounce homogenizer (15-20 strokes). Pellet nuclei.
- Collagenase/Hyaluronidase Digestion (for fibrous tissues): Resuspend pellet in 1 mL of digestion buffer (RPMI + 1 mg/mL Collagenase IV + 30 U/mL Hyaluronidase). Incubate 30 min at 37°C with gentle rotation.
- Nuclear Lysis: Proceed with standard nuclear lysis in LB3 (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-Deoxycholate, 0.5% N-lauroylsarcosine) supplemented with 0.1% SDS. Sonicate using a focused ultrasonicator (Covaris S220) with intensified conditions: 20% Duty Factor, 200 PIP, 200 cycles/burst for 10-12 minutes.
- Chromatin Clarification: Centrifuge at 20,000 x g for 15 min at 4°C to remove insoluble debris and lipid layers.
Table 1: Optimized Sonication Conditions for Difficult Tissues
| Tissue Type | Recommended Sonication Device | Duty Factor | PIP | Cycles/Burst | Time | Goal Fragment Size |
|---|---|---|---|---|---|---|
| Cardiac Muscle | Focused Ultrasonicator | 20% | 200 | 200 | 12-15 min | 200-500 bp |
| Adipose/Brain | Focused Ultrasonicator | 15% | 180 | 200 | 10 min + Lipid Clean-up | 200-500 bp |
| Fibrotic Tumor | Focused Ultrasonicator + Enzymatic Pre-digestion | 22% | 220 | 200 | 12 min | 200-500 bp |
| Standard Culture Cells | Bath or Focused Ultrasonicator | 10% | 140 | 200 | 8-10 min | 200-500 bp |
Protocol for FFPE Samples
FFPE chromatin is crosslinked, fragmented, and damaged, requiring reversal of formalin crosslinks and specialized repair steps prior to ChIP.
Detailed Methodology: FFPE Chromatin Extraction & Repair
- Deparaffinization & Rehydration: Cut 5-10 x 10 µm FFPE sections. Incubate in xylene (2 x 10 min), then in a graded ethanol series (100%, 95%, 70%, 50% - 5 min each). Rinse in PBS.
- Crosslink Reversal & Proteinase K Digestion: Suspend in 1 mL of Digest Buffer (100 mM Tris pH 8.0, 10 mM EDTA, 0.5% SDS) with 1 µL of Proteinase K (20 mg/mL). Incubate 4 hrs at 65°C, then add another 1 µL Proteinase K and incubate overnight at 65°C.
- Chromatin Extraction: Cool, then add an equal volume of Phenol:Chloroform:Isoamyl Alcohol. Centrifuge. Transfer aqueous phase and precipitate DNA with glycogen and ethanol.
- Chromatin Shearing: Resuspend pellet in TE buffer. Sonicate using Covaris S220 to ~200-600 bp fragments (validate on Bioanalyzer). Note: Sonication is for size selection, not crosslink reversal.
- DNA End-Repair & Ligation: Use a high-efficiency library preparation kit (e.g., NEB Next Ultra II) designed for damaged DNA, which includes end-repair, dA-tailing, and adapter ligation steps that also serve to "repair" the FFPE chromatin substrate.
- ChIP: Proceed with standard ChIP protocol using the repaired, adapter-ligated chromatin. Use magnetic beads for cleanup steps.
Table 2: Impact of FFPE Repair Steps on ChIP-seq Data Quality
| Protocol Step | Key Metric | Unoptimized Protocol | Optimized (with Repair) | Measurement Method |
|---|---|---|---|---|
| Chromatin Extraction | DNA Yield (per 10µm section) | 50 - 200 ng | 500 ng - 2 µg | Qubit dsDNA HS Assay |
| Post-Sonication | % of Fragments in 200-600 bp range | 20-40% | 60-80% | TapeStation/Bioanalyzer |
| Post-ChIP | Library Complexity (Non-Redundant Reads) | 1-3 million | 8-15 million | Picard Tools EstimateLibraryComplexity |
| Mapping | Mapping Rate to Reference Genome | 40-60% | 75-90% | Bowtie2/BWA output |
| Background | Fraction of Reads in Peaks (FRiP) | 0.5-1% | 5-15% | MACS2/SPP |
Protocol for Rare Cell Populations (<10,000 cells)
The principal challenge is signal loss from nonspecific adsorption and low statistical power. Strategies focus on maximal recovery and amplification.
Detailed Methodology: Microfluidic or Carrier-Assisted ChIP
Option A: Microfluidic (µChIP)
- Cell Lysis: Directly load sorted cells into a microfluidic chamber (e.g., FluiChip platform).
- On-Chip Processing: Perform all steps—lysis, sonication (via acoustic shearing), immunoprecipitation, and washes—within nanoliter-scale volumes on the integrated chip. This minimizes surface area and volume, reducing losses.
- Elution & Purification: Elute chromatin in a minimal volume (5-10 µL) and transfer directly to library prep.
Option B: Carrier-Assisted ChIP
- Carrier Addition: Mix 5,000-10,000 target cells with 50,000-100,000 Drosophila melanogaster S2 cells (or other phylogenetically distant carrier cells) prior to crosslinking. Ensure antibody does not recognize carrier chromatin.
- Standard ChIP: Perform crosslinking and standard ChIP protocol. The carrier provides bulk chromatin, normalizing bead handling and wash losses.
- Bioinformatic Separation: During analysis, map reads to a combined (human + Drosophila) reference genome. Discard Drosophila reads; analyze human reads only.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Challenging Sample ChIP-seq
| Reagent / Kit Name | Vendor (Example) | Function in Protocol | Key Benefit for Challenging Samples |
|---|---|---|---|
| Covaris cryoPREP | Covaris | Automated cryogenic tissue pulverization | Standardizes input from heterogeneous, tough tissues. |
| Chromatin Extraction Kit for FFPE | Active Motif / Diagenode | Optimized buffers for FFPE chromatin extraction & repair | Maximizes yield of usable chromatin from archives. |
| NEBNext Ultra II FS DNA Library Prep | New England Biolabs | Includes enzymes for fragmented/damaged DNA input | Ideal for FFPE and low-input repair & adapter ligation. |
| FluiChip µPAC | Fluigent | Microfluidic chip for nanoliter-scale reactions | Minimizes volume, dramatically reducing loss in rare-cell ChIP. |
| Drosophila S2 Cells | Invitrogen | Carrier chromatin for low-input ChIP | Provides bulk for efficient IP, bioinformatically separable. |
| Methylcellulose | Sigma-Aldrich | Viscosity agent added to ChIP wash buffers | Reduces bead loss during washing steps in low-input protocols. |
| Protein A/G Magnetic Beads | Thermo Fisher / Millipore | Solid-phase for antibody capture | Lower non-specific binding vs. agarose, better for low-input. |
| Th5 Transposase (Tagmentation) | Illumina (Nextera) | Simultaneous fragmentation and adapter tagging | Reduces steps, improving yield from limited cells. |
Experimental Workflow & Pathway Diagrams
Title: Workflow for Challenging Sample ChIP-seq Protocols
Title: FFPE Chromatin Repair Pathway for ChIP-seq
Title: Rare Cell ChIP-seq Strategy Decision Tree
Integrating these protocol adaptations into the standard ChIP-seq workflow, as outlined in the overarching thesis, dramatically expands the reach of epigenomic research. By systematically addressing the unique challenges of difficult tissues, FFPE samples, and rare cells, researchers can generate robust data from previously intractable sample types. This is paramount for translational studies in oncology, neurology, and rare diseases, where such samples are often the only available source of biological material for epigenetic drug target discovery and biomarker development.
Application Note: Ensuring Library Integrity in ChIP-seq for Epigenomics Research
Within the context of a comprehensive thesis on ChIP-seq for epigenomics research, robust Quality Control (QC) is paramount. The reliability of downstream sequencing data and biological interpretation hinges on the precise assessment of ChIP-enriched DNA samples and constructed sequencing libraries. This document details the application and protocols for three critical, complementary QC checkpoints: Agarose Gel Electrophoresis, Bioanalyzer/TapeStation analysis, and quantitative PCR (qPCR) Pre-Sequencing. These stages evaluate fragment size distribution, library concentration, and enrichment efficiency, respectively, forming an essential triad for successful epigenomic profiling.
Detailed Experimental Protocols
Agarose Gel Electrophoresis for ChIP-DNA Fragment Assessment
Purpose: To visually confirm successful fragmentation of crosslinked chromatin (post-sonication) and to estimate the size distribution of immunoprecipitated DNA prior to library construction.
Materials:
- ChIP-eluted DNA (or sonicated input DNA).
- Standard agarose, molecular biology grade.
- 1X TAE or TBE electrophoresis buffer.
- DNA loading dye (6X).
- DNA molecular weight ladder (e.g., 100 bp ladder).
- Nucleic acid stain (e.g., SYBR Safe, GelRed).
- Gel electrophoresis system and power supply.
- Gel imaging system (UV or blue light).
Protocol:
- Prepare a 1.5-2% agarose gel by dissolving agarose in 1X electrophoresis buffer. Cool to ~55°C, add nucleic acid stain as per manufacturer's instructions, and cast the gel.
- Mix 5-10 µL of ChIP-DNA or input DNA with 1/6 volume of 6X loading dye.
- Load the samples alongside an appropriate DNA ladder.
- Run the gel at 5-8 V/cm in 1X electrophoresis buffer until the dye front has migrated sufficiently.
- Image the gel. Expected Result: A smear centered between 100-500 bp for optimally sonicated chromatin. A discrete, low-molecular-weight band (~100 bp) indicates over-sonication, while high-molecular-weight smears suggest under-sonication.
Bioanalyzer/TapeStation Analysis for Sequencing Library QC
Purpose: To obtain precise, high-resolution fragment size distribution and molar concentration of the final ChIP-seq library before sequencing. This step is critical for accurate pooling and sequencing cluster density optimization.
Materials:
- Final, amplified ChIP-seq library.
- Agilent High Sensitivity DNA Kit (e.g., Agilent 2100 Bioanalyzer) or D1000/High Sensitivity D1000 ScreenTape (Agilent TapeStation).
- Appropriate reagents and chips/ladders as specified by the kit.
Protocol (Bioanalyzer Example):
- Prepare the gel-dye mix and priming station as detailed in the Agilent High Sensitivity DNA Assay Guide.
- Load 1 µL of marker into the appropriate ladder and sample wells.
- Dilute 1 µL of the ChIP-seq library in 5 µL of deionized water. Load 1 µL of this dilution into a sample well.
- Vortex the chip for 1 minute at 2400 rpm.
- Run the chip on the Agilent 2100 Bioanalyzer instrument.
- Data Analysis: The software generates an electrophoretogram and a virtual gel image. Key outputs include the average library size (bp) and the library molar concentration (nM). The profile should show a clear, single peak corresponding to the insert size plus adapters, with minimal adapter-dimer contamination (~120-130 bp).
qPCR Pre-Sequencing (Pre-Seq) Enrichment Validation
Purpose: To quantitatively verify the specific enrichment of target regions in the ChIP sample versus a control (Input DNA) prior to the costly sequencing step.
Materials:
- Diluted ChIP-DNA and Input DNA (pre-library construction, or from a test aliquot of the library).
- Primer pairs for positive control regions (known binding sites for the target protein) and negative control regions (genomic regions not bound by the target protein, e.g., gene deserts, inactive promoters).
- SYBR Green qPCR Master Mix.
- Real-time PCR instrument and plates/tubes.
Protocol:
- Dilute all DNA samples to a uniform concentration (e.g., 0.1-1 ng/µL).
- Prepare qPCR reactions in triplicate for each sample-primer combination: 1X SYBR Green Master Mix, forward/reverse primers (e.g., 200 nM each), and template DNA.
- Run qPCR with a standard cycling program: 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min, with a melt curve analysis.
- Calculate the average Ct value for each triplicate. Determine the percent input or fold enrichment.
- % Input Method: % Input = 2^(Ct[Input] - Ct[ChIP]) * F * 100, where F is the Input dilution factor.
- Fold Enrichment: Fold Enrichment = 2^(Ct[ChIPnegative] - Ct[ChIPpositive]) when comparing the same sample across different primers.
Expected Result: Significant enrichment (e.g., >10-fold) at positive control regions compared to negative control regions validates a successful ChIP experiment.
Data Presentation
Table 1: Comparative Summary of QC Checkpoints in ChIP-seq Workflow
| Checkpoint | Sample Stage | Key Metrics Assessed | Typical Acceptable Range | Purpose in Thesis Context |
|---|---|---|---|---|
| Agarose Gel | Post-sonication ChIP-DNA | Fragment size distribution | Smear: 100-500 bp (centered ~200-300 bp) | Confirm proper chromatin shearing; essential for mapping resolution. |
| Bioanalyzer | Final sequencing library | Peak size, concentration, adapter-dimer % | Size: Target insert + adapters; [ ]: >2 nM; Adapters: <10% | Ensure proper library construction, accurate pooling, and optimal sequencing. |
| qPCR Pre-Seq | Pre-library ChIP-DNA or test library | Fold enrichment, % Input | >10-fold enrichment at positive vs. negative sites | Validate biological specificity of the immunoprecipitation prior to sequencing investment. |
Mandatory Visualization
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for ChIP-seq QC
| Item | Function in QC | Key Consideration for Thesis Research |
|---|---|---|
| High Sensitivity DNA Assay (Bioanalyzer/TapeStation) | Provides precise, automated sizing and quantification of limited DNA samples like ChIP libraries. Enables accurate pooling of multiplexed libraries. | The high sensitivity range (5-500 pg/µL) is essential for quantifying low-yield ChIP-seq libraries without wasting material. |
| SYBR Safe/GelRed DNA Stain | A safer, non-mutagenic alternative to ethidium bromide for visualizing DNA fragments on agarose gels. | Allows for rapid, in-lab assessment of chromatin shearing efficiency post-sonication and post-library amplification. |
| SYBR Green qPCR Master Mix | Enables quantitative assessment of target enrichment via real-time PCR. Sensitive and cost-effective for Pre-Seq validation. | Must be used with validated, locus-specific primer sets for positive and negative control genomic regions relevant to the epigenomic target. |
| High Sensitivity DNA Ladder | Provides precise size reference for both agarose gels and capillary electrophoresis systems. | Critical for accurate size determination of sheared chromatin and final library inserts, which impacts sequencing data analysis. |
| SPRIselect/AMPure XP Beads | Used for size-selective purification of DNA to remove primers, adapter-dimers, and unwanted large fragments. | A critical step post-library PCR to clean up the final library before Bioanalyzer analysis and sequencing. |
Validating ChIP-seq Data and Comparing Epigenomic Profiling Techniques
1. Introduction Within a ChIP-seq-based epigenomics thesis, post-sequencing quality control (QC) is the critical gateway to reliable biological interpretation. This step determines if the raw sequencing data is of sufficient quality to proceed with peak calling, motif analysis, and downstream epigenomic profiling. Assessing key metrics and mapping rates directly impacts the validity of conclusions regarding transcription factor binding or histone modification landscapes in drug discovery contexts.
2. Core Sequencing Metrics: Definitions and Thresholds The initial QC involves evaluating the raw sequencing output using tools like FastQC and MultiQC. Key metrics are summarized below.
Table 1: Essential Post-Sequencing QC Metrics and Interpretation
| Metric | Optimal Range/Value | Implication of Deviation | Common Cause in ChIP-seq |
|---|---|---|---|
| Per Base Sequence Quality (Phred Score) | ≥ Q30 for majority of cycles | High error rate, unreliable base calls. | Degraded library, cluster density issues on flow cell. |
| Per Sequence Quality Scores | Mean ≥ Q30 | Overall read quality is poor. | Systematic sequencing run issue. |
| Adapter Content | ≤ 2-5% overall | Significant data loss after trimming, shorter inserts. | Library fragment size shorter than read length. |
| Overrepresented Sequences | None (or known controls) | PCR duplication, adapter dimers, or contamination. | Over-amplification during library prep, insufficient size selection. |
| GC Content | Matches organism/distribution | Contamination from other species or sequences. | PCR bias, or presence of a dominant contaminant. |
3. Protocol: Comprehensive Post-Sequencing QC Workflow
Protocol 3.1: Initial Raw Read Assessment with FastQC/MultiQC Objective: To generate a comprehensive quality report for single or multiple ChIP-seq libraries. Materials: Raw FASTQ files, High-performance computing (HPC) cluster or local server with Conda environment. Procedure:
- Environment Setup: Install tools via Conda:
conda create -n qc fastqc multiqc -c bioconda -c conda-forge - Run FastQC: For each FASTQ file, execute:
fastqc sample_R1.fastq.gz -o ./fastqc_reports/ - Aggregate Reports: Navigate to the reports directory and run:
multiqc .This generates a singlemultiqc_report.html. - Interpretation: Open the HTML report. Systematically check each metric in Table 1. Pay special attention to adapter content and per-base quality to inform trimming parameters.
Protocol 3.2: Read Trimming and Filtering with Trim Galore! Objective: To remove adapter sequences, poor-quality bases, and low-quality reads. Materials: FASTQ files from 3.1, Trim Galore! (wrapper for Cutadapt and FastQC). Procedure:
- Run Trim Galore: Execute with standard parameters, adjusting for adapter type:
trim_galore --paired sample_R1.fastq.gz sample_R2.fastq.gz --output_dir ./trimmed/ --cores 4 - QC Post-trimming: Run FastQC/MultiQC (Protocol 3.1) on the trimmed
*_val_*.fq.gzfiles to confirm improvement. - Output: High-quality, adapter-free FASTQ files ready for alignment.
4. Assessing Mapping Rates and Alignment-Specific Metrics Mapping rate is the percentage of quality-filtered reads that align uniquely or non-uniquely to the reference genome. It is a primary indicator of ChIP enrichment and sample quality.
Table 2: Mapping Metrics and Their Significance in ChIP-seq
| Metric | Target (Typical Mammalian ChIP-seq) | Rationale & Impact on Analysis |
|---|---|---|
| Overall Alignment Rate | > 80-90% of trimmed reads | Low rates suggest poor library complexity, contamination, or wrong reference genome. |
| Uniquely Mapped Rate | > 70-80% of trimmed reads | High multi-mapping reads can complicate peak calling. |
| ChIP-seq Fraction of Reads in Peaks (FRiP) | 1-5% (Input); >5-30% (Enriched TF); >10-30% (Histone Marks) | Key signal-to-noise metric. Low FRiP indicates poor enrichment. |
| Duplicate Rate (PCR duplicates) | < 20-50% (library-dependent) | Very high rates indicate low complexity, limiting detection of rare binding events. |
Protocol 3.3: Read Alignment and Metric Calculation using Bowtie2 and SAMtools Objective: To map reads to a reference genome and compute alignment statistics. Materials: Trimmed FASTQs, reference genome index (e.g., GRCh38/hg38), Bowtie2, SAMtools, Picard Tools. Procedure:
- Alignment: Align paired-end reads:
bowtie2 -x /path/to/genome_index -1 sample_R1_val_1.fq.gz -2 sample_R2_val_2.fq.gz -S sample_aligned.sam --threads 8 - SAM to BAM Conversion:
samtools view -bS sample_aligned.sam -o sample_aligned.bam - Sort BAM File:
samtools sort sample_aligned.bam -o sample_sorted.bam - Calculate Basic Mapping Stats:
samtools flagstat sample_sorted.bam > sample_flagstat.txt - Mark/Remove Duplicates (using Picard):
picard MarkDuplicates I=sample_sorted.bam O=sample_deduped.bam M=sample_dup_metrics.txt REMOVE_DUPLICATES=true - Index Final BAM:
samtools index sample_deduped.bam - Extract Metrics: Review
flagstat.txtanddup_metrics.txtto populate Table 2 metrics.
Title: Post-Sequencing and Mapping QC Workflow for ChIP-seq Data
5. The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for ChIP-seq Post-Sequencing QC & Analysis
| Item / Solution | Function in Post-Sequencing QC | Example / Notes |
|---|---|---|
| FastQC Software | Provides initial visual report on sequencing quality metrics (Phred scores, GC content, adapter contamination). | Open-source tool; run locally or on a cluster. |
| Trim Galore! / Cutadapt | Automates adapter trimming and removal of low-quality bases based on FastQC results. | Critical for removing sequencing artifacts before alignment. |
| Reference Genome (FASTA) & Index | The sequence against which reads are aligned to determine origin. Mapping rate depends on correct reference. | Ensembl/GENCODE genome builds (e.g., GRCh38.p13). Must be indexed for aligner (Bowtie2/BWA). |
| Alignment Software (Bowtie2/BWA) | Performs the alignment of sequencing reads to the reference genome, outputting SAM/BAM files. | Bowtie2 is widely used for its speed and sensitivity in ChIP-seq. |
| SAMtools/Picard Toolkit | Utilities for processing SAM/BAM files: sorting, indexing, marking duplicates, and extracting metrics. | samtools flagstat gives mapping rates; Picard calculates duplicate rates. |
| DeepTools | Suite for advanced QC visualization post-alignment (read coverage, correlation plots, FRiP calculation). | plotFingerprint command assesses enrichment quality. |
| High-Performance Computing (HPC) Resource | Essential for running alignment and QC tools on large sequencing datasets efficiently. | Local servers or cloud-based solutions (AWS, Google Cloud). |
Title: Key Metrics Determine ChIP-seq Data Fate
Within the context of a thesis on ChIP-seq protocols for epigenomics research, the selection and parameterization of peak calling algorithms are critical steps that directly impact downstream biological interpretations. Peak calling identifies genomic regions where protein-DNA interactions, such as transcription factor binding or histone modifications, are enriched. This note details the application, protocols, and parameter optimization for two widely used algorithms: MACS2 (Model-based Analysis of ChIP-Seq) and SICER (Spatial Clustering Approach for the Identification of ChIP-Enriched Regions).
MACS2
MACS2 is designed primarily for pinpoint protein factors (e.g., transcription factors) with sharp, localized peaks. It employs a dynamic Poisson distribution to model the background tag distribution, incorporates a shift size to better locate the precise binding site, and calculates a false discovery rate (FDR).
SICER
SICER is optimized for diffuse histone marks (e.g., H3K36me3, H3K9me3) that produce broad enrichment regions. It uses a clustering approach that accounts for spatial information, allowing it to identify significantly enriched genomic islands by accounting for gaps within clusters.
Table 1: Core Algorithm Characteristics and Typical Use Cases
| Feature | MACS2 | SICER |
|---|---|---|
| Primary Design | Sharp peaks (Transcription Factors) | Broad domains (Histone Modifications) |
| Statistical Model | Dynamic Poisson / Negative Binomial | Randomization and Poisson |
| Key Strength | High resolution for precise binding sites | Sensitivity to widespread, diffuse signals |
| Critical Parameter | --qvalue (FDR cutoff), --extsize |
windowSize, gapSize, FDR |
| Typical Output | NarrowPeak files (point-source peaks) | BroadPeak/Island files (enriched regions) |
Parameter Optimization for Sensitivity and Specificity
Tuning parameters is essential to balance sensitivity (true positive rate) and specificity (true negative rate). Incorrect parameters can lead to false discoveries or missed genuine binding events.
Table 2: Key Parameters and Their Impact on Sensitivity/Specificity
| Algorithm | Parameter | Default | Effect of Increasing Value | Impact on Sensitivity | Impact on Specificity |
|---|---|---|---|---|---|
| MACS2 | --qvalue |
0.05 | Stricter significance threshold | Decreases | Increases |
--extsize |
Estimated | User-defined fragment extension size | Context-dependent | Context-dependent | |
--broad |
Off | Enables broad peak calling | Increases for broad marks | May decrease | |
| SICER | windowSize (W) |
200 bp | Larger scanning window | Decreases for sharp peaks | May increase for broad marks |
gapSize (G) |
600 bp | Allowed gap between windows | Increases (merges islands) | May decrease | |
FDR |
0.01 | Stricter false discovery cutoff | Decreases | Increases |
Detailed Experimental Protocols
Protocol 1: Standard MACS2 Peak Calling for Transcription Factor ChIP-seq
This protocol is for analyzing a transcription factor (TF) ChIP-seq dataset with a corresponding control (e.g., Input DNA).
Materials & Reagents:
- FASTQ Files: Raw sequencing reads for ChIP and Control samples.
- Reference Genome: Species-specific genome assembly (e.g., GRCh38, mm10).
- Alignment Software: Bowtie2 or BWA.
- MACS2 Software: Installed via conda (
conda install -c bioconda macs2).
Procedure:
- Alignment: Align ChIP and control FASTQ files to the reference genome. Convert output SAM to BAM, sort, and index.
Peak Calling with MACS2: Run MACS2 in standard narrow peak calling mode.
Output Interpretation: Primary outputs are
*_peaks.narrowPeak(BED format) and*_peaks.xls(summary table).
Protocol 2: SICER Peak Calling for Broad Histone Marks
This protocol is for identifying broad domains from histone mark ChIP-seq data.
Materials & Reagents:
- Processed BAM Files: Sorted, aligned reads for ChIP and Control.
- SICER Software: Available as a Python package (
pySICERor standalone). - BedTools: For file format manipulations.
Procedure:
- BAM to BED Conversion: Convert input BAM files to BED format.
Run SICER: Execute SICER with parameters optimized for broad marks.
Output Interpretation: The key file is
*-islands-summary, listing significant genomic islands.
Visualizing the Peak Calling Workflow and Decision Logic
Diagram Title: Decision Workflow for Selecting Peak Calling Algorithm and Key Parameters
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Reagents for ChIP-seq Peak Calling Analysis
| Item | Function/Description | Example Product/Software |
|---|---|---|
| NGS Sequencing Platform | Generates raw sequencing reads (FASTQ) from immunoprecipitated DNA. | Illumina NovaSeq, NextSeq |
| Alignment Tool | Maps sequencing reads to a reference genome. | Bowtie2, BWA, STAR |
| Peak Calling Software | Identifies statistically enriched genomic regions. | MACS2, SICER, HOMER |
| Genome Assembly | Reference sequence for alignment and annotation. | UCSC hg38, ENSEMBL GRCh38 |
| Control Sample | Input DNA or IgG ChIP; essential for background noise modeling. | Sheared genomic DNA |
| High-Performance Computing (HPC) | Computational resource for processing large NGS datasets. | Local cluster, Cloud (AWS, GCP) |
| Genomic Annotation Database | Provides biological context to called peaks (e.g., nearest gene). | ENSEMBL, UCSC RefSeq |
| Visualization Software | Allows inspection of peak signals across the genome. | IGV, UCSC Genome Browser |
Application Notes
This document details the essential validation experiments required to confirm findings from a primary ChIP-seq analysis within an epigenomics research thesis. Relying solely on bioinformatic peaks can lead to false positives due to antibody non-specificity, sequencing artifacts, or peak-calling errors. A tripartite validation strategy—quantitative ChIP (qChIP), Western Blot, and de novo motif analysis—provides orthogonal, experimental confirmation of protein-DNA interactions, target protein expression, and biological relevance.
- qChIP validates the enrichment observed in ChIP-seq at specific loci, moving from genome-wide data to focused, quantitative assessment.
- Western Blot confirms the specificity of the ChIP-grade antibody and the presence of the target protein (and its relevant modification) in the sample.
- Motif Analysis determines if the enriched genomic regions contain known or novel binding sequences, linking the interaction to a plausible molecular mechanism.
The integration of these methods significantly strengthens the thesis conclusions, ensuring robustness for downstream applications in target discovery and drug development.
Protocols
1. Quantitative Chromatin Immunoprecipitation (qChIP) Protocol
- Objective: To validate the enrichment of specific genomic regions identified in ChIP-seq.
- Materials: Crosslinked cell pellets, ChIP-validated antibody, Protein A/G magnetic beads, sonicator, qPCR system, primers for positive control, negative control, and 3-5 candidate peaks.
- Method:
- Chromatin Preparation: Follow the primary ChIP-seq protocol to sonicate crosslinked chromatin to 200-500 bp fragments.
- Immunoprecipitation: For each test antibody and IgG control, incubate 2-5 µg chromatin with 1-5 µg antibody overnight at 4°C. Add beads for 2 hours.
- Wash & Elution: Wash beads with low-salt, high-salt, LiCl, and TE buffers. Elute chromatin in fresh elution buffer.
- Reverse Crosslinks: Incubate eluates and input samples at 65°C overnight.
- DNA Purification: Use a PCR purification kit.
- qPCR Analysis: Run samples in triplicate. Calculate % Input for each region:
% Input = 100 * 2^(Ct[Input] - Ct[IP]). Compare enrichment in specific antibody IP vs. IgG control.
2. Western Blot Validation of ChIP Antibody
- Objective: To confirm antibody specificity and target protein expression/modification in the sample.
- Materials: Whole cell extract from same sample used for ChIP, SDS-PAGE gel, transfer apparatus, ChIP antibody, secondary HRP-antibody, ECL substrate.
- Method:
- Sample Preparation: Lyse cells in RIPA buffer with protease/phosphatase inhibitors.
- Electrophoresis & Transfer: Load 20-50 µg protein, separate by SDS-PAGE, transfer to PVDF membrane.
- Blocking & Incubation: Block with 5% non-fat milk. Incubate with ChIP antibody (1:1000 dilution) overnight at 4°C.
- Detection: Incubate with appropriate HRP-conjugated secondary antibody (1:5000). Develop with ECL reagent. The blot should show a single band at the expected molecular weight.
3. De Novo Motif Discovery & Analysis Protocol
- Objective: To identify enriched DNA binding motifs within ChIP-seq peaks.
- Materials: FASTA file of peak sequences (top 500-1000 peaks by p-value), computational tools (HOMER, MEME-ChIP).
- Method (Using HOMER):
- Prepare Sequences:
findMotifsGenome.pl <peak file.bed> <reference genome> <output directory> -size 200 - Run De Novo Discovery: The command above performs de novo motif finding. Results include known motif comparisons.
- Analysis: Review output files
knownResults.txtandhomerResults.html. A successful ChIP will show significant enrichment for the known motif of the target protein and/or a novel, conserved sequence.
- Prepare Sequences:
Data Presentation
Table 1: qChIP Validation Data for Hypothetical Transcription Factor "X"
| Genomic Region | IgG % Input (Mean ± SD) | α-TFX % Input (Mean ± SD) | Fold Enrichment (α-TFX/IgG) | Validated? |
|---|---|---|---|---|
| Positive Control (Known Site) | 0.05 ± 0.01 | 2.50 ± 0.30 | 50.0 | Yes |
| Peak 1 (Chr5:120,450,100) | 0.04 ± 0.01 | 1.80 ± 0.20 | 45.0 | Yes |
| Peak 2 (Chr12:88,200,750) | 0.06 ± 0.02 | 1.65 ± 0.15 | 27.5 | Yes |
| Negative Control (Gene Desert) | 0.03 ± 0.01 | 0.07 ± 0.02 | 2.3 | No |
Table 2: De Novo Motif Analysis Summary (HOMER)
| Motif Logo (Top 3) | p-value (Best Match) | % of Targets with Motif | Known Match |
|---|---|---|---|
| (ATTSGCGCCAAT) | 1e-25 | 42% | NF-κB (p65) |
| (TGANTCA) | 1e-18 | 28% | AP-1 (c-Jun/c-Fos) |
| (GGGCGG) | 1e-12 | 15% | SP/KLF Family |
Mandatory Visualizations
Title: Tripartite ChIP-seq Validation Workflow
Title: Step-by-Step qChIP Protocol for Validation
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Validation |
|---|---|
| ChIP-Validated Antibody | Primary reagent for immunoprecipitation; must be validated for specificity and efficacy in ChIP applications. |
| Protein A/G Magnetic Beads | For efficient capture of antibody-chromatin complexes, enabling rapid washing. |
| Crosslinking Reagent (e.g., 1% Formaldehyde) | Fixes protein-DNA interactions in vivo prior to chromatin shearing. |
| Sonicator (Covaris or tip-based) | Shears chromatin to optimal fragment size (200-500 bp) for resolution. |
| qPCR Master Mix & Validated Primers | For precise quantification of DNA enrichment at specific loci post-ChIP. |
| ChIP-seq Grade Cell Line/Tissue | Biological material with documented expression of target protein/epitope. |
| De Novo Motif Discovery Software (HOMER/MEME) | Bioinformatics tools to identify enriched DNA sequence motifs in peak regions. |
| Modified Nucleotide Analogs (Optional) | For spike-in normalization (e.g., Drosophila chromatin) in quantitative experiments. |
Application Notes
Within the broader thesis on optimizing ChIP-seq for epigenomics research, the emergence of CUT&Tag represents a paradigm shift in mapping protein-DNA interactions. This analysis compares these two cornerstone techniques across critical operational parameters, providing a framework for researchers to select the appropriate method based on project goals, sample type, and resource constraints. CUT&Tag, leveraging a Protein A-Tn5 transposase fusion protein targeted by antibodies, fundamentally reduces background and input requirements compared to the crosslinking, sonication, and precipitation steps of traditional ChIP-seq.
Quantitative Comparison
Table 1: Performance Comparison of ChIP-seq and CUT&Tag
| Parameter | Chromatin Immunoprecipitation Sequencing (ChIP-seq) | Cleavage Under Targets and Tagmentation (CUT&Tag) |
|---|---|---|
| Typical Cell Input | 0.5 – 10 million cells | 500 – 100,000 cells |
| Hands-on Time | 2-4 days | ~1 day |
| Sequencing Depth | 20-50 million high-quality reads | 1-10 million high-quality reads |
| Background Noise | Higher (crosslinking/sonication artifacts) | Very low (in situ tagmentation) |
| Resolution | 50-300 bp (dependent on sonication) | Single-nucleotide (transposase insertion sites) |
| Throughput (Cells to Libraries) | Lower (multi-day protocol) | Higher (potentially one-day protocol) |
| Primary Cost Driver | Sequencing depth, antibodies, reagents | Antibodies, commercial kits |
| Best For | Robust protocols, histone marks, abundant TFs, frozen tissues. | Low-input samples, sensitive cells, high-throughput profiling, delicate co-factors. |
Experimental Protocols
Protocol 1: Standard Crosslinking ChIP-seq for Histone Marks (from Thesis Framework)
- Day 1: Crosslinking & Cell Lysis. Treat cells with 1% formaldehyde for 10 min at room temperature. Quench with 125 mM glycine. Wash cells with cold PBS. Lyse cells in LB1 (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100) for 10 min on ice. Pellet nuclei, then lyse in LB2 (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) for 10 min on ice.
- Day 1-2: Chromatin Shearing. Pellet nuclei, resuspend in Sonication Buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris-HCl pH 8.0). Sonicate chromatin to ~200-500 bp fragments using a focused ultrasonicator (e.g., Covaris). Confirm fragment size by agarose gel electrophoresis. Clarify lysate by centrifugation.
- Day 2: Immunoprecipitation. Dilute chromatin in ChIP Dilution Buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH 8.0, 167 mM NaCl). Pre-clear with Protein A/G beads for 1 hour. Incubate supernatant with 1-5 µg of specific antibody overnight at 4°C with rotation.
- Day 3: Bead Capture & Washes. Add blocked Protein A/G beads for 2 hours. Wash beads sequentially: once with Low Salt Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 150 mM NaCl), once with High Salt Wash Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 500 mM NaCl), once with LiCl Wash Buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH 8.0), and twice with TE Buffer.
- Day 3: Elution & Decrosslinking. Elute chromatin twice with Elution Buffer (1% SDS, 0.1 M NaHCO3). Add NaCl to 200 mM and reverse crosslinks overnight at 65°C.
- Day 4: DNA Purification & Library Prep. Treat with RNase A and Proteinase K. Purify DNA using SPRI beads. Construct sequencing libraries using a commercial kit (e.g., NEBNext Ultra II DNA Library Prep Kit) with appropriate size selection.
Protocol 2: CUT&Tag for Transcription Factors
- Step 1: Cell Preparation & Permeabilization. Harvest 100,000 cells. Wash twice in Wash Buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, 1x Protease Inhibitor). Resuspend in Digitonin-containing Wash Buffer (0.01% Digitonin) and incubate 10 min on ice. Wash once in Digitonin Buffer.
- Step 2: Primary Antibody Binding. Resuspend cells in 50 µL Antibody Buffer (Digitonin Wash Buffer + 2 mM EDTA + 0.1% BSA). Add primary antibody (1:50-1:100 dilution). Incubate overnight at 4°C with rotation.
- Step 3: Secondary Antibody Binding. Wash twice with 1 mL Digitonin Buffer to remove unbound primary antibody. Resuspend in 50 µL Digitonin Buffer containing 1:100 dilution of Guinea Pig anti-Rabbit (or appropriate) secondary antibody. Incubate for 1 hour at room temperature.
- Step 4: pA-Tn5 Binding & Tagmentation. Wash twice with Digitonin Buffer. Resuspend in 50 µL Digitonin Buffer containing a 1:100 dilution of pre-loaded pA-Tn5 transposase (commercially available). Incubate for 1 hour at room temperature.
- Step 5: Activation & DNA Extraction. Wash twice with Digitonin Buffer. Resuspend in 100 µL Tagmentation Buffer (10 mM MgCl2 in Digitonin Buffer). Incubate at 37°C for 1 hour. Add 10 µL of 0.5 M EDTA, 3 µL of 10% SDS, and 2.5 µL of 20 mg/mL Proteinase K. Incubate at 55°C for 1 hour to stop reaction and release DNA.
- Step 6: Library Amplification. Purify DNA directly using SPRI beads. Amplify libraries using 1-2 µL of purified DNA in a 20-50 µL PCR reaction with barcoded primers and a high-fidelity polymerase (e.g., NEBNext HiFi 2X PCR Master Mix). Purify final library with SPRI beads and quantify.
Mandatory Visualization
Diagram Title: ChIP-seq vs CUT&Tag Experimental Workflow Comparison
Diagram Title: Decision Tree for Choosing ChIP-seq or CUT&Tag
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for ChIP-seq and CUT&Tag
| Item | Function | Example/Catalog Consideration |
|---|---|---|
| High-Specificity Antibody | Binds the target protein/epitope of interest. Critical for both methods. | Validated ChIP/CUT&Tag-grade antibodies (e.g., from Cell Signaling, Abcam, Diagenode). |
| Protein A/G Magnetic Beads | (ChIP-seq) Captures antibody-protein-DNA complexes. | Dynabeads Protein A/G, Sera-Mag beads. |
| pA-Tn5 Fusion Protein | (CUT&Tag) Core enzyme; Protein A binds antibody, Tn5 tagments DNA. | Commercial kits (e.g., EpiCypher, Cell Signaling, Active Motif). |
| Digitonin | (CUT&Tag) Gently permeabilizes the cell membrane while leaving the nuclear envelope intact. | High-purity digitonin solutions. |
| Formaldehyde (37%) | (ChIP-seq) Reversible crosslinker to fix protein-DNA interactions. | Molecular biology grade, methanol-free. |
| Tn5 Transposase | (CUT&Tag, also for library prep) Enzyme that simultaneously fragments and tags DNA with adapters. | Illumina Nextera/ATM, homemade loaded Tn5. |
| SPRI Magnetic Beads | Size selection and purification of DNA fragments post-IP or tagmentation. | Beckman Coulter AMPure XP, homemade SPRI beads. |
| High-Fidelity PCR Mix | Amplifies libraries post-tagmentation (CUT&Tag) or after adapter ligation (ChIP-seq). | NEBNext HiFi 2X PCR Master Mix, KAPA HiFi HotStart. |
| Dual-Indexed Adapters | For multiplexing samples during high-throughput sequencing. | Illumina TruSeq, IDT for Illumina, NEBNext Multiplex Oligos. |
Within the framework of a comprehensive thesis on ChIP-seq protocol for epigenomics research, understanding chromatin accessibility is paramount. While ChIP-seq identifies protein-DNA interactions, ATAC-seq maps open chromatin regions. These techniques are not redundant but provide complementary, multi-dimensional views of chromatin state, essential for researchers and drug development professionals seeking to understand gene regulation mechanisms.
Core Principles and Comparative Data
Table 1: Core Comparison of ChIP-seq (for Accessibility Factors) and ATAC-seq
| Feature | ChIP-seq (for e.g., H3K27ac, H3K4me3) | ATAC-seq (Assay for Transposase-Accessible Chromatin) |
|---|---|---|
| Primary Target | Protein-DNA interactions (histone modifications, transcription factors). | Nucleosome-free, accessible DNA regions. |
| Biological Insight | Indirect inference of accessibility via active/poised enhancer/promoter marks. | Direct mapping of physical chromatin accessibility. |
| Resolution | 100-300 bp (defined by sonication/fragment size). | Single-nucleotide resolution (due to Tn5 insertion). |
| Starting Material | High (500k-1M cells for histone marks; >50k for TFs). | Low (500-50,000 cells, with robust protocols for <100). |
| Key Reagent | Target-specific antibody. | Hyperactive Tn5 transposase. |
| Protocol Duration | 3-5 days (crosslinking, sonication, IP). | ~3 hours (from cells to sequencing library). |
| Primary Data | Enrichment peaks at protein-binding sites. | Insertion sites defining accessible chromatin. |
| Challenge | Antibody specificity and availability; crosslinking artifacts. | Mitochondrial DNA contamination; data complexity from nucleosome positioning. |
Table 2: Quantitative Output Metrics from a Typical Integrative Study
| Data Type | Typical Peak Number (Human Genome) | Concordance Rate (Overlap) | Unique Information Provided |
|---|---|---|---|
| ATAC-seq Peaks | 80,000 - 150,000 | 70-85% of sites overlap with active histone marks. | De novo accessibility sites, nucleosome positions. |
| H3K27ac ChIP-seq Peaks | 50,000 - 100,000 | ~80% colocalize with ATAC-seq peaks. | Active enhancer and promoter identification. |
| H3K4me3 ChIP-seq Peaks | 25,000 - 50,000 | ~90% colocalize with ATAC-seq peaks. | Active promoter identification. |
| Unique ATAC-seq-only sites | 10,000 - 30,000 | N/A | Potential poised/regulatory elements without canonical marks. |
Detailed Experimental Protocols
Protocol 1: Standard ChIP-seq for Histone Marks (H3K27ac)
This protocol is a core component of the overarching thesis on ChIP-seq for epigenomics.
I. Cell Crosslinking and Harvesting
- Add 1% formaldehyde directly to culture medium. Incubate for 10 min at room temperature (RT) with gentle agitation.
- Quench crosslinking by adding 125 mM glycine for 5 min at RT.
- Wash cells twice with ice-cold PBS. Pellet cells and flash-freeze pellet or proceed.
II. Chromatin Preparation and Shearing
- Lyse cells in SDS Lysis Buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH 8.1) with protease inhibitors.
- Sonicate lysate to shear chromatin to 200-500 bp fragments. Use a focused ultrasonicator (e.g., Covaris) for reproducibility.
- Clarify sonicate by centrifugation. Dilute supernatant 10-fold in ChIP Dilution Buffer.
III. Immunoprecipitation and Washing
- Pre-clear chromatin with Protein A/G beads for 1-2 hours at 4°C.
- Incubate supernatant with 2-5 µg of validated anti-H3K27ac antibody overnight at 4°C.
- Add pre-blocked Protein A/G beads for 2 hours.
- Wash beads sequentially: Low Salt Wash Buffer (once), High Salt Wash Buffer (once), LiCl Wash Buffer (once), TE Buffer (twice).
IV. Elution, Reverse Crosslinking, and Purification
- Elute chromatin twice with Fresh Elution Buffer (1% SDS, 0.1M NaHCO3).
- Add 5M NaCl and reverse crosslink at 65°C overnight.
- Treat with RNase A and Proteinase K.
- Purify DNA using SPRI bead-based clean-up.
V. Library Preparation and Sequencing
- Use a commercial library preparation kit for Illumina platforms.
- Perform end-repair, A-tailing, and adapter ligation on immunoprecipitated DNA.
- Amplify library with 12-18 PCR cycles.
- Size-select (250-350 bp) and sequence on an Illumina platform (≥20 million reads).
Protocol 2: Standard ATAC-seq
This complementary protocol provides direct accessibility data.
I. Cell Preparation and Lysis
- Harvest 50,000 viable cells. Wash once with cold PBS.
- Lyse cells in cold Lysis Buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Igepal CA-630) for 3 min on ice.
- Immediately pellet nuclei at 500 x g for 10 min at 4°C. Resuspend pellet in 50 µL of Transposition Mix.
II. Transposition Reaction
- Prepare 50 µL Transposition Mix: 25 µL 2x TD Buffer, 2.5 µL Tn5 Transposase (Illumina), 22.5 µL nuclease-free water, 0.5-1.0 µL 1% Digitonin (for permeabilization).
- Incubate the reaction at 37°C for 30 min in a thermomixer with shaking (1000 rpm).
- Purify transposed DNA immediately using a MinElute PCR Purification Kit or SPRI beads. Elute in 21 µL Elution Buffer.
III. Library Amplification and Purification
- Amplify library using 2x KAPA HiFi HotStart ReadyMix and 1.25 µM of custom Ad1 and Ad2.x barcoded primers.
- Run a 5-cycle pre-PCR: 72°C for 5 min, 98°C for 30 sec; then 5 cycles of [98°C for 10 sec, 63°C for 30 sec, 72°C for 1 min].
- Pause, remove 5 µL for qPCR side reaction to determine additional cycles (Cq). Calculate remaining cycles (N = round( (Cq - 4) / 2 ) ).
- Resume PCR for N more cycles. Purify final library with SPRI beads (0.6x ratio). Sequence on Illumina (≥50 million paired-end reads).
Visualizing the Complementary Workflow and Data Integration
Title: Complementary ChIP-seq and ATAC-seq Workflow Integration
Title: Data Integration Logic for Complementary Analysis
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for ChIP-seq and ATAC-seq Studies
| Reagent/Material | Function | Example/Note |
|---|---|---|
| Validated ChIP-grade Antibodies | Specific immunoprecipitation of target protein or histone modification. | Anti-H3K27ac (abcam ab4729), Anti-H3K4me3 (CST 9751). Critical for ChIP-seq specificity. |
| Hyperactive Tn5 Transposase | Simultaneously fragments and tags accessible chromatin with sequencing adapters. | Illumina Tagmentase TDE1, or custom loaded/DIY Tn5. Core of ATAC-seq. |
| Magnetic Protein A/G Beads | Efficient capture of antibody-chromatin complexes for washing and elution. | Invitrogen Dynabeads. Reduce non-specific background in ChIP. |
| SPRI (Solid Phase Reversible Immobilization) Beads | Size-selective purification and clean-up of DNA fragments post-IP or tagmentation. | Beckman Coulter AMPure XP. Used in both protocols for consistent cleanup. |
| Cell Permeabilization Reagent | Enhances Tn5 access to nuclear chromatin in ATAC-seq. | Digitonin. Used in the "OMNI-ATAC" protocol for improved signal. |
| Dual-Indexed Adapter Kits | Allows multiplexing of samples during NGS library preparation for cost-efficiency. | Illumina TruSeq, Nextera XT. Essential for pooling ChIP-seq/ATAC-seq libraries. |
| Sonication Device (Covaris) | Provides reproducible, controlled acoustic shearing of crosslinked chromatin for ChIP-seq. | Covaris S220. Preferable over bath sonication for uniform fragment size. |
| High-Sensitivity DNA Assay Kits | Accurate quantification of low-concentration libraries prior to sequencing. | Agilent Bioanalyzer/TapeStation, Qubit dsDNA HS Assay. |
Integrating ChIP-seq with RNA-seq and Hi-C for Systems Biology Insights
Application Notes
This application note details a unified experimental and computational framework for integrating ChIP-seq, RNA-seq, and Hi-C data to derive systems-level insights into gene regulatory mechanisms. This integrated approach is central to a broader thesis on advancing ChIP-seq protocols for epigenomics research, moving beyond singular assay analysis to a multi-dimensional understanding of how transcription factor binding, chromatin state, 3D architecture, and gene expression coordinately drive cellular function and disease.
Core Insights:
- Mechanistic Linkage: Establishes causal relationships between transcription factor (TF) or histone mark localization (ChIP-seq), chromatin looping (Hi-C), and transcriptional output (RNA-seq).
- Enhancer-Gene Assignment: Combines ChIP-seq peaks (e.g., H3K27ac) with Hi-C contact maps to confidently link distal regulatory elements to their target promoters.
- Context for Differential Expression: Provides a regulatory context for differentially expressed genes (RNA-seq) by overlaying changes in TF binding, promoter/enhancer activity, and topologically associating domain (TAD) integrity.
- Disease Mechanism Elucidation: Identifies non-coding genetic variants (e.g., GWAS SNPs) that disrupt TF binding motifs (ChIP-seq), alter chromatin contacts (Hi-C), and lead to aberrant gene expression (RNA-seq) in complex diseases.
Key Quantitative Integrative Metrics: Table 1: Key Quantitative Metrics from Multi-Omics Integration
| Metric | Data Source | Typical Range/Value | Interpretation | ||
|---|---|---|---|---|---|
| Peak-to-Gene Link Score | ChIP-seq + Hi-C | 0 to 1 (probability) | Confidence that a distal ChIP-seq peak contacts a gene promoter. | ||
| Expression-Fold Change | RNA-seq | Log2FC > | 1 | , adj. p < 0.05 | Significant up/down-regulation of a gene. |
| Differential Contact Strength | Hi-C (e.g., HiChIP) | Log2FC in contact frequency | Significant increase/decrease in chromatin looping. | ||
| Co-localization P-value | ChIP-seq peaks (multiple factors) | -log10(p) > 3 (p < 0.001) | Statistical significance of spatial overlap between two TF binding sites. | ||
| TAD Boundary Shift | Hi-C | Shift > 50 kb between conditions | Major reorganization of 3D chromatin architecture. |
Detailed Protocols
Protocol 1: Sequential Multi-Omic Profiling from a Single Biological Sample Goal: Generate ChIP-seq, RNA-seq, and Hi-C (or HiChIP) data from the same cell population to minimize biological variability.
- Cell Crosslinking: Crosslink ~1x10^7 cells with 1% formaldehyde for 10 min at RT. Quench with 125 mM glycine.
- Nuclear Extraction & Aliquotting: Lyse cells and isolate nuclei. Split nuclei into three aliquots.
- Aliquot A (ChIP-seq): Sonicate chromatin to 200-500 bp fragments. Perform immunoprecipitation with target antibody (e.g., H3K4me3, CTCF). Reverse crosslinks, purify DNA.
- Aliquot B (RNA-seq): Isolate total RNA using TRIzol. Perform poly-A selection or rRNA depletion. Prepare stranded cDNA library.
- Aliquot C (Hi-C/HiChIP): Digest chromatin with a 4-cutter restriction enzyme (e.g., MboI). Fill ends and mark with biotin-dCTP. Perform proximity ligation. For HiChIP, include an immunoprecipitation step post-ligation with an antibody (e.g., H3K27ac). Shear DNA, pull down biotinylated ligation junctions, and prepare library.
- Sequencing: Sequence ChIP-seq and RNA-seq libraries on Illumina NovaSeq (50M and 30M paired-end reads, respectively). Sequence Hi-C/HiChIP library on NovaSeq (400M+ paired-end reads).
Protocol 2: Computational Integration Pipeline Goal: Integrate datasets to identify coordinated regulatory changes.
- Primary Analysis:
- ChIP-seq: Align reads (Bowtie2), call peaks (MACS2), and annotate peaks (HOMER).
- RNA-seq: Align reads (STAR), quantify gene expression (featureCounts), perform differential expression (DESeq2).
- Hi-C: Process reads (HiC-Pro or Juicer), generate contact matrices, identify TADs (Arrowhead algorithm), and call loops (HiCCUPS).
- Integration:
- Regulatory Network Inference: Use a tool like GRaNIE or PECA to statistically integrate TF peaks, ATAC-seq accessibility, and RNA-seq expression to build cell-type-specific gene regulatory networks.
- Enhancer-Gene Linking: Use PeakHi-C or ChIP-Interaction to assign distal ChIP-seq peaks to target genes via Hi-C contact maps. Filter for correlations with gene expression.
- 3D Change Analysis: Visualize differential ChIP-seq signals (e.g., ΔH3K27ac) on differential Hi-C contact maps (using pyGenomeTracks) to identify alterations in active chromatin hubs.
Visualizations
Title: Multi-Omic Data Generation & Integration Workflow
Title: Integrating 1D Signals & 3D Contacts for Regulation
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Integrated Multi-Omics
| Reagent / Kit | Function in Protocol | Key Feature |
|---|---|---|
| Formaldehyde (1-3%) | Reversible crosslinking of protein-DNA and protein-protein interactions. | Preserves in vivo chromatin architecture for ChIP-seq and Hi-C. |
| Magna ChIP Protein A/G Beads | Immunoprecipitation of chromatin-antibody complexes. | High specificity, low background for ChIP-seq and HiChIP. |
| NEBNext Ultra II DNA Library Prep Kit | Preparation of sequencing-ready libraries from ChIP DNA. | High-efficiency adapter ligation for low-input samples. |
| Illumina TruSeq Stranded mRNA Kit | Preparation of RNA-seq libraries from poly-A RNA. | Strand-specific information preserves directionality. |
| Arima-HiC Kit | Optimized reagent suite for Hi-C library preparation. | Simplified, high-resolution protocol with high ligation efficiency. |
| Diagenode MicroPlex Library Preparation Kit v3 | Library prep for very low input ChIP-seq/ATAC-seq. | Ideal for scarce clinical samples in multi-omic studies. |
| CTCF or H3K27ac Antibody (ChIP-seq grade) | Target-specific immunoprecipitation. | Validated for ChIP-seq and HiChIP; crucial for data quality. |
| Dynabeads MyOne Streptavidin C1 Beads | Pulldown of biotinylated Hi-C ligation junctions. | Efficient recovery of chimeric contacts for Hi-C sequencing. |
Conclusion
A robust ChIP-seq protocol is the cornerstone of modern epigenomics, enabling precise mapping of the regulatory genome. Mastering the foundational principles, meticulous execution of the crosslinking and immunoprecipitation steps, proactive troubleshooting, and rigorous bioinformatic validation are all critical for generating biologically meaningful data. As the field evolves, ChIP-seq remains a gold standard, but its integration with newer, lower-input techniques like CUT&Tag and multi-omics approaches will further propel discovery. For biomedical and clinical researchers, high-quality ChIP-seq data directly fuels the identification of disease-associated regulatory variants, mechanisms of drug action, and novel epigenetic therapeutic targets, bridging the gap between chromatin biology and translational medicine.