Mastering ChIP-seq Analysis for Transcription Factors: A Complete Guide for Biomedical Researchers
This comprehensive guide provides researchers and drug development professionals with a complete workflow for ChIP-seq data analysis focused on transcription factor binding.
Mastering ChIP-seq Analysis for Transcription Factors: A Complete Guide for Biomedical Researchers
Abstract
This comprehensive guide provides researchers and drug development professionals with a complete workflow for ChIP-seq data analysis focused on transcription factor binding. Covering everything from foundational principles to advanced optimization, the article details experimental design, quality control, peak calling, downstream bioinformatics analysis, troubleshooting common issues, and validation strategies. Readers will gain practical knowledge for accurately identifying TF binding sites, interpreting functional genomic data, and applying these insights to understand gene regulation in health and disease contexts.
Understanding Transcription Factor ChIP-seq: From Experimental Design to Raw Data
Within a comprehensive ChIP-seq data analysis workflow for transcription factor (TF) research, understanding the fundamental distinctions between TF and histone mark ChIP-seq is critical. These differences dictate experimental design, data processing, and biological interpretation. This guide delineates the unique challenges and considerations specific to TF ChIP-seq, contrasting them with the more stable nature of histone mark profiling.
Core Biological and Technical Distinctions
The inherent properties of TFs versus histone modifications create divergent experimental landscapes.
Key Comparison Table
| Feature | Transcription Factor (TF) ChIP-seq | Histone Mark ChIP-seq |
|---|---|---|
| Target Stability | Transient, dynamic binding (seconds-minutes). | Stable, cumulative modification (hours-days). |
| Binding Site Resolution | Sharp, narrow peaks (~100-500 bp). | Broad, diffuse regions (1-10 kb for some marks). |
| Cross-linking Requirement | Mandatory (formaldehyde). | Often optional (native ChIP possible). |
| Antibody Specificity | Extremely high; concerns about epitope masking. | Generally high; many well-validated antibodies. |
| Signal-to-Noise Ratio | Typically lower, with high background. | Typically higher, with clear enrichment. |
| Peak Calling Challenge | Precise summit identification critical. | Defining region boundaries is key. |
| Required Sequencing Depth | High (20-50 million reads). | Moderate to high (10-40 million reads). |
| Primary Biological Question | Identification of specific cis-regulatory elements. | Mapping chromatin state and domain organization. |
Detailed Methodological Considerations
Experimental Protocol: TF ChIP-seq with Formaldehyde Cross-linking
Objective: To capture transient, protein-DNA interactions in vivo. Procedure:
- Cross-linking: Treat cells with 1% formaldehyde for 8-12 minutes at room temperature. Quench with 125 mM glycine.
- Cell Lysis & Sonication: Lyse cells in SDS buffer. Shear chromatin via sonication to 200-500 bp fragments. Critical: Optimize sonication to avoid over/under-fragmentation.
- Immunoprecipitation: Incubate lysate with protein-specific antibody (e.g., anti-TF antibody) conjugated to magnetic beads overnight at 4°C. Use species-matched IgG as control.
- Washing & Elution: Wash beads stringently (e.g., low salt, high salt, LiCl, TE buffers). Elute complexes with fresh elution buffer (1% SDS, 0.1M NaHCO3).
- Reverse Cross-linking & Purification: Incubate eluate at 65°C overnight with high salt to reverse cross-links. Treat with RNase A and Proteinase K. Purify DNA via column-based methods.
- Library Preparation & Sequencing: Construct sequencing libraries from purified DNA (end-repair, A-tailing, adapter ligation, PCR amplification). Sequence on an appropriate platform (e.g., Illumina).
Experimental Protocol: Histone Mark ChIP-seq (Native)
Objective: To map stable epigenetic modifications. Procedure:
- Micrococcal Nuclease (MNase) Digestion: Isolate nuclei. Digest chromatin with MNase to yield primarily mononucleosomes. Note: Formaldehyde cross-linking can be used but is often omitted.
- Chromatin Extraction & IP: Extract chromatin in low-salt buffer. Immunoprecipitate with histone modification-specific antibody (e.g., anti-H3K4me3) overnight.
- Washing, Elution, & DNA Purification: Wash, elute, and purify DNA as in steps 4-5 of TF protocol, omitting reverse cross-linking if native.
- Library Preparation & Sequencing: Proceed as in TF protocol.
Visualizing the Workflow Divergence
TF vs Histone ChIP-seq Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
| Reagent/Material | Function | Key Consideration |
|---|---|---|
| Formaldehyde (37%) | Reversible protein-DNA cross-linking. | Critical for TFs. Optimize time/temp to capture transient interactions without masking epitopes. |
| MNase | Digests linker DNA for native histone ChIP. | Used for nucleosome-level mapping in histone ChIP; less common in TF ChIP. |
| Magnetic Protein A/G Beads | Solid support for antibody capture. | Choice of A/G depends on antibody species/isotype. Consistency is key for reproducibility. |
| High-Specificity Primary Antibodies | Binds target antigen (TF or histone mark). | TF ChIP: Validate for ChIP; epitope may be cross-link sensitive. Histone: Many commercial, validated options exist. |
| Protease Inhibitor Cocktail | Preserves protein integrity during lysis/IP. | Essential in all steps prior to reverse cross-linking. |
| Glycine | Quenches formaldehyde cross-linking reaction. | Stops cross-linking to prevent over-fixation and epitope damage. |
| Proteinase K | Digests proteins post-IP to release DNA. | Required after reverse cross-linking in TF protocols. |
| SPRI/AMPure Beads | Size-selects and purifies DNA fragments. | Used in library prep and post-IP clean-up. More consistent than column-based methods. |
| Sequencing Adapters & Indexes | Enables multiplexed, high-throughput sequencing. | Use unique dual indexes to reduce index hopping artifacts. |
| Control Antibodies (IgG, Input) | Determines non-specific background. | IgG: Species-matched. Input: Non-immunoprecipitated, sheared chromatin. Both are mandatory for robust analysis. |
Data Analysis Implications
The distinctions above cascade into the analysis workflow. TF ChIP-seq requires sophisticated background modeling for narrow peak calling (e.g., with MACS2). Motif discovery within peaks is a primary downstream analysis. Histone mark data often employs broader peak callers or segmentation algorithms (e.g., ChromHMM) to define chromatin states, with emphasis on read density profiles across genomic features.
Analysis Parameter Table
| Analysis Step | TF ChIP-seq Priority | Histone Mark ChIP-seq Priority |
|---|---|---|
| Read Alignment | Remove duplicates cautiously (may lose signal). | Often aggressive duplicate removal. |
| Peak Calling | Model local background; focus on summit. | Use broad peak settings; focus on region. |
| Control Subtraction | Absolute reliance on control (IgG/Input). | Input control highly important. |
| Downstream Analysis | De novo motif discovery, pathway analysis. | Chromatin state annotation, gene body plots. |
Successful ChIP-seq analysis for transcription factor research hinges on recognizing its unique demands: the imperative of cross-linking, the battle against low signal-to-noise, the need for high-resolution peak detection, and the absolute requirement for rigorously validated antibodies. These factors collectively differentiate it from the more tractable analysis of histone modifications and must be accounted for at every stage, from experimental design through computational interpretation, within a robust ChIP-seq workflow thesis.
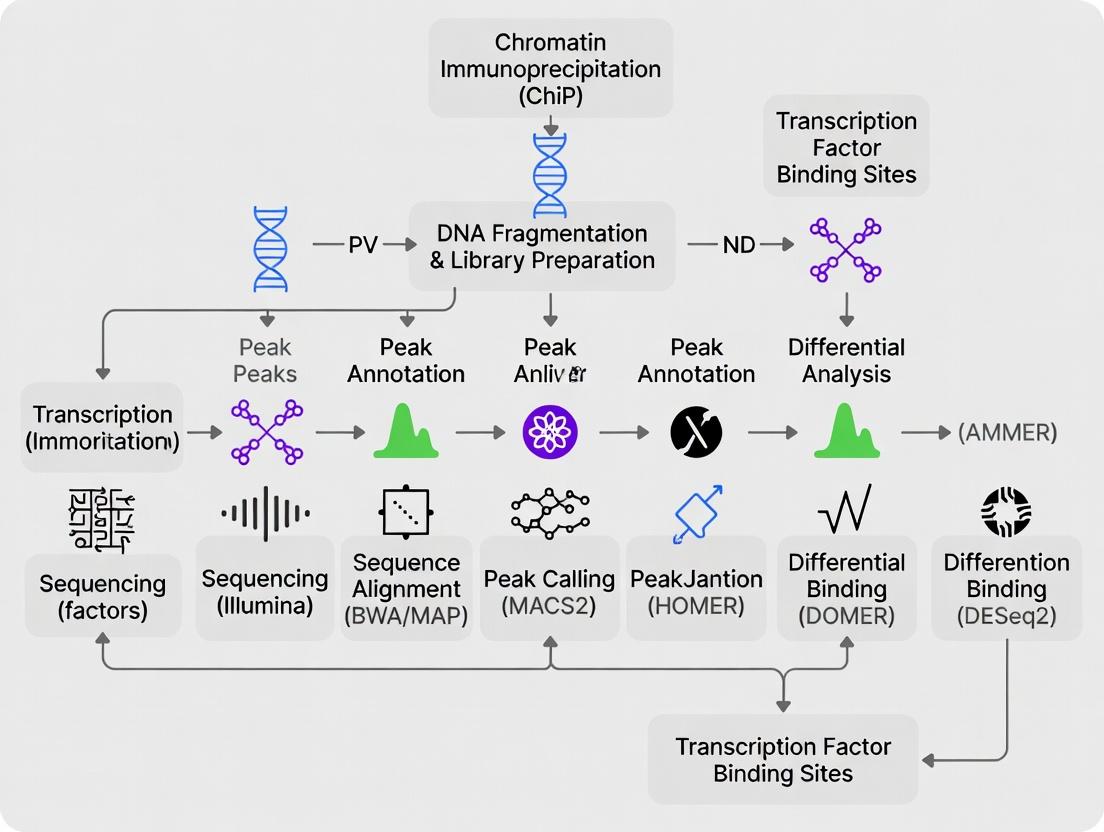
This technical guide details the core experimental pillars of the Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) workflow, framed within a broader thesis on establishing a robust pipeline for transcription factor (TF) research and data analysis. The quality of the final genomic data and subsequent biological interpretation is fundamentally dependent on the rigor applied in these initial wet-lab stages.
Chemical Crosslinking
Crosslinking captures transient, protein-DNA interactions by creating covalent bonds. For TFs, which bind DNA with high specificity but relatively low stability, this is a critical first step.
Protocol: Formaldehyde Crosslinking for Adherent Cells
- Grow cells to 70-80% confluence.
- Add 37% formaldehyde directly to culture media to a final concentration of 1%. Gently swirl.
- Incubate at room temperature (RT) for 8-12 minutes with gentle agitation.
- Quench the reaction by adding glycine to a final concentration of 0.125 M. Incubate for 5 minutes at RT.
- Aspirate media, wash cells twice with ice-cold phosphate-buffered saline (PBS).
- Scrape cells into PBS containing protease inhibitors. Pellet cells (500 x g, 4°C, 5 min). Cell pellets can be flash-frozen and stored at -80°C.
Table 1: Comparison of Common Crosslinkers for ChIP-seq
| Crosslinker | Target | Spacer Arm | Primary Use in ChIP | Key Consideration |
|---|---|---|---|---|
| Formaldehyde | Primary amines (Lys); DNA-protein, protein-protein | ~2 Å | Standard for TFs, co-factors | Rapid, reversible; may under-crosslink heterochromatin. |
| DSG (Disuccinimidyl glutarate) | Primary amines (protein-protein) | ~7.7 Å | Often used prior to formaldehyde (sequential) | Stabilizes protein complexes before DNA-protein fixation. |
| EGS (Ethylene glycol bis(succinimidyl succinate)) | Primary amines (protein-protein) | ~16.1 Å | Sequential crosslinking for difficult targets | Longer spacer can help capture larger complexes. |
Diagram: Formaldehyde Crosslinking of TF-DNA Complex
Chromatin Shearing via Sonication
Following crosslinking and nuclei isolation, chromatin must be fragmented to an optimal size (200-600 bp) to achieve sufficient resolution while maintaining protein-DNA complex integrity.
Protocol: Ultrasonic Sonication (Covaris-focused Acoustics)
Equipment: Covaris S220 or equivalent, milliTUBE (130µl). Starting Material: ~1 million fixed nuclei, resuspended in 130µl shearing buffer (1% SDS, 10mM EDTA, 50mM Tris-HCl pH 8.0). Covaris Settings:
- Peak Incident Power: 140 W
- Duty Factor: 5%
- Cycles per Burst: 200
- Treatment Time: 8-12 minutes
- Temperature: Maintained at 4-6°C
- Expected Output: Majority of fragments between 200-500 bp.
Table 2: Shearing Method Comparison
| Method | Principle | Fragment Range | Consistency | Throughput | |
|---|---|---|---|---|---|
| Ultrasonic (Covaris) | Focused acoustic energy | Tunable (100-1000 bp) | High, reproducible | Medium (1 sample/run) | |
| Bath Sonicator | Cavitation in water bath | Broad, less tunable | Moderate, user-dependent | High (multi-sample) | |
| Enzymatic (MNase) | Digests linker DNA | Mononucleosome (~147 bp) | High for nucleosome studies | High | Not suitable for most TFs. |
Diagram: Chromatin Shearing and Quality Control Workflow
Antibody Selection and Validation
The antibody is the single most critical reagent in ChIP-seq. Its specificity directly defines the signal-to-noise ratio of the experiment.
Protocol: Pre-Immunoprecipitation Antibody Validation
- Western Blot: Perform on crosslinked and sheared chromatin (reversed) and whole cell extract. Confirm a single band at the expected molecular weight.
- Immunofluorescence: Confirm expected subcellular localization (nuclear for TFs).
- Peptide Competition: Pre-incubate antibody with its target antigenic peptide. Successful competition should abolish the ChIP signal.
- Use of Knockout/Knockdown Controls: ChIP in a cell line where the target TF is genetically ablated. A valid antibody should yield no significant peaks.
- Comparative Genomic Enrichment (CGE): Compare peak profiles and enrichment at known positive control loci with a well-characterized antibody.
Table 3: Antibody Source and Validation Criteria
| Criteria | Polyclonal | Monoclonal | Validation Recommendation |
|---|---|---|---|
| Specificity | May recognize multiple epitopes; risk of off-target binding. | Single epitope; higher specificity. | Must pass both WB on crosslinked material and KO control validation. |
| Affinity | High, due to multivalent binding. | Consistent, but may be lower. | Compare enrichment (% input) at a positive locus vs. IgG control (>10-fold). |
| Lot Consistency | Variable between immunizations. | Highly consistent. | Request lot-specific validation data from vendor. |
| Common Source | Rabbit, goat. | Rabbit, mouse, rat. | Prefer vendors participating in ABR (Antibody Registry). |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Materials for Core ChIP-seq Experimental Steps
| Reagent/Material | Function | Key Consideration |
|---|---|---|
| UltraPure Formaldehyde (37%) | Reversible crosslinking agent. | Use fresh, methanol-free aliquots for consistent efficiency. |
| Protease Inhibitor Cocktail (PIC) | Prevents proteolytic degradation of TFs/complexes during cell lysis. | Must be added fresh to all buffers prior to lysis and IP. |
| Covaris milliTUBE (130µl) | AFA fiber tube for optimal acoustic energy transfer during shearing. | Ensure no air bubbles are present in the sample. |
| Dynabeads Protein A/G | Magnetic beads for antibody immobilization and complex pulldown. | Choose A, G, or A/G mix based on host species of ChIP antibody. |
| RNAse A & Proteinase K | Enzymes for digesting RNA and proteins during crosslink reversal & DNA purification. | Critical for clean, high-yield DNA recovery post-IP. |
| SPRI/AMPure XP Beads | Solid-phase reversible immobilization beads for size selection and DNA clean-up. | Ratio of beads to sample determines fragment size selection. |
| High-Specificity ChIP-grade Antibody | Binds specifically to the target protein of interest. | The critical reagent. Non-negotiable requirement for validated, ChIP-seq-grade antibodies. |
| Control IgG (Species-matched) | Negative control for non-specific antibody binding. | Must be from the same host species as the ChIP antibody. |
| SYBR Green qPCR Master Mix | For quantitative PCR validation of enrichment at known sites pre-sequencing. | Test 3-5 positive and negative control genomic loci. |
Diagram: Antibody Validation Decision Pathway
The interdependent steps of crosslinking, sonication, and antibody selection form the non-negotiable foundation of any ChIP-seq experiment for transcription factors. Rigorous optimization and validation at each stage, guided by the quantitative benchmarks and protocols herein, are prerequisites for generating high-fidelity data that can withstand rigorous bioinformatic analysis and yield biologically meaningful insights into gene regulatory mechanisms.
Within the comprehensive workflow of ChIP-seq data analysis for transcription factor (TF) research, the integrity of biological conclusions rests upon robust experimental controls. Three controls are non-negotiable: the Input DNA control, the IgG negative control, and properly designed biological replicates. This guide details their essential functions, implementation, and analysis within a modern ChIP-seq framework.
The Role and Execution of Core Controls
Input DNA Control
Function: The Input control consists of genomic DNA that has been crosslinked and fragmented but not subjected to immunoprecipitation. It accounts for biases in sequencing arising from genomic DNA accessibility, local chromatin structure, PCR amplification, and sequencing efficiency.
Detailed Protocol:
- Take an aliquot of the crosslinked, sonicated chromatin sample (typically 1-10% of the volume used for a single IP).
- Reverse crosslinks by adding NaCl to a final concentration of 200 mM and incubating at 65°C for a minimum of 4 hours (or overnight).
- Purify DNA using a standard phenol-chloroform extraction or a silica-membrane-based kit.
- Process this purified DNA in parallel with the IP samples through end-repair, adapter ligation, and PCR amplification for sequencing library construction.
IgG Negative Control
Function: This control uses a non-specific immunoglobulin G (IgG) from the same host species as the specific antibody in a parallel immunoprecipitation. It identifies regions of the genome that are non-specifically enriched due to protein-protein or protein-DNA interactions with the bead matrix or the Fc region of antibodies.
Detailed Protocol:
- Use the same chromatin preparation as for the specific TF antibody IP.
- Substitute the specific antibody with an equivalent mass (usually 1-5 µg) of non-specific IgG (e.g., rabbit IgG for a rabbit polyclonal TF antibody).
- Perform the entire IP, wash, elution, and reverse crosslinking procedure identically to the test sample.
- Process the purified DNA for sequencing alongside the specific IP and Input samples.
Biological Replicates
Function: Biological replicates are independent chromatin preparations from separate cell cultures or tissue samples. They account for stochastic biological variability, allowing researchers to distinguish reproducible binding events from technical noise and random background.
Detailed Protocol:
- Design: Perform at least two (ideally three) independent cell harvests, chromatin preparations, and immunoprecipitations on different days.
- Independence: Maintain cell cultures separately. For tissues, use samples from different animals or pooled from multiple dissections.
- Processing: Replicates should be processed identically but can be multiplexed with unique barcodes during library preparation and sequenced across different lanes to avoid batch effects.
Table 1: Recommended Sequencing Depth and Replicates for TF ChIP-seq
| Control / Sample Type | Minimum Recommended Sequencing Depth (Reads) | Minimum Number of Biological Replicates | Primary Purpose in Analysis |
|---|---|---|---|
| Transcription Factor IP | 20 - 50 million | 3 | Identify binding peaks |
| Input DNA | Matched to or greater than deepest IP sample | Matched to IP replicates | Background normalization |
| IgG Control | 20 - 50 million | At least 1 | Assess non-specific binding |
Table 2: Impact of Controls on Peak Calling Metrics (Typical Values)
| Analysis Scenario | Number of Peaks Called | False Discovery Rate (FDR) | Reproducibility (IDR*) Score |
|---|---|---|---|
| TF-IP vs. Input DNA | ~15,000 | 1-5% | 0.05 - 0.10 |
| TF-IP vs. IgG | ~8,000 | 1-5% | 0.05 - 0.15 |
| TF-IP vs. Input & IgG (combined model) | ~12,000 | <1% | <0.05 |
| TF-IP without control | >40,000 | >25% | >0.30 |
*IDR: Irreproducible Discovery Rate. Lower is better.
Visualizing the Control Framework in ChIP-seq
Diagram 1: ChIP-seq Control Integration from Experiment to Analysis
The Scientist's Toolkit: Essential Reagent Solutions
Table 3: Key Research Reagents for ChIP-seq Controls
| Reagent / Material | Function & Importance | Example Product/Catalog |
|---|---|---|
| Non-specific Species-Matched IgG | Critical for the IgG control IP. Must match the host species and isotype (e.g., Rabbit IgG) of the primary antibody. | Millipore Sigma, 12-370 |
| Protein A/G Magnetic Beads | For antibody capture. High binding capacity and low non-specific DNA binding are essential for clean IgG controls. | Thermo Fisher, 10002D |
| Formaldehyde (37%) | For crosslinking protein-DNA interactions. Must be fresh for consistent crosslinking efficiency across replicates. | Thermo Fisher, 28906 |
| Glycine (2.5M Solution) | To quench crosslinking, stopping the reaction uniformly across all samples. | Thermo Fisher, J22638 |
| Chromatin Shearing Reagent (Sonicator) | For consistent DNA fragmentation (200-500 bp). Calibrated sonication is vital for reproducible IPs. | Covaris, S220 |
| DNA Clean & Concentrator Kit | For purifying DNA after reverse crosslinking. High recovery and purity are needed for sensitive library prep. | Zymo Research, D4033 |
| High-Sensitivity DNA Assay Kit | To accurately quantify low-concentration ChIP DNA before library construction (e.g., Qubit dsDNA HS Assay). | Thermo Fisher, Q32851 |
| Unique Dual-Indexed Library Prep Kit | Allows multiplexing of biological replicates and controls, reducing batch effects and cost. | Illumina, 20020495 |
| SPRIselect Beads | For size selection and clean-up during library prep. Provides reproducible fragment size selection. | Beckman Coulter, B23318 |
Within the broader ChIP-seq data analysis workflow for transcription factor research, the initial step of understanding raw sequencing data is fundamental. This guide provides an in-depth technical examination of FASTQ files and the quality metrics essential for downstream analysis.
The Structure of a FASTQ File
A FASTQ file is the primary raw data output from high-throughput sequencing platforms (e.g., Illumina). It stores both the nucleotide sequence and its corresponding per-base quality scores. Each sequence read is represented by a block of four lines:
- Line 1 (Header): Begins with
@, followed by a unique sequence identifier and optional metadata (instrument, run ID, flowcell lane, coordinates). - Line 2 (Sequence): The raw nucleotide sequence (A, C, G, T, N).
- Line 3 (Separator): Begins with
+, sometimes followed by the same identifier as line 1 (optional). - Line 4 (Quality String): Encodes the per-base quality score for each nucleotide in Line 2 using ASCII characters.
Quality Score Encoding: Phred Scale
Quality scores (Q-scores) predict the probability (P) of a base call being incorrect. The relationship is defined as: Q = -10 × log₁₀(P). Two major encodings exist, differing by an ASCII offset:
Table 1: Common Quality Score Encodings
| Encoding Format | ASCII Offset | Quality Score Range (Q) | Typical Sequencing Platform |
|---|---|---|---|
| Sanger / Illumina 1.8+ | 33 | 0 to 93 | Illumina (post-2011), PacBio, Ion Torrent |
| Illumina 1.3+ / 1.5+ | 64 | 0 to 62 | Illumina (ca. 2008-2011) |
For example, in Sanger format (offset 33), a quality character "F" (ASCII 70) corresponds to Q = 70 - 33 = 37. This means P(error) ≈ 10⁻³·⁷ ≈ 0.0002, or a base call accuracy of 99.98%.
Essential Quality Control Metrics and Tools
For ChIP-seq experiments targeting transcription factors, high-quality reads are critical to identify precise binding sites. Initial Quality Control (QC) is performed using tools like FastQC and MultiQC.
Table 2: Key FASTQ Quality Metrics for ChIP-seq QC
| Metric | Ideal Outcome for TF ChIP-seq | Potential Issue Indicated |
|---|---|---|
| Per Base Sequence Quality | Q ≥ 30 across all cycles. | Degradation towards read ends suggests loss of sequencing fidelity. |
| Per Sequence Quality Scores | Sharp peak at high Q (≥30). | Broad/low peak indicates many low-quality reads. |
| Sequence Duplication Levels | Low duplication rate for standard ChIP-seq. | High duplication may indicate low library complexity or PCR over-amplification. |
| Adapter Content | Near 0% contamination. | Presence of adapter sequences indicates short fragment reads that require trimming. |
| GC Content | Matches organism's genomic GC% (~40% for human, ~50% for D. melanogaster). | Deviation may indicate contamination or biased fragmentation. |
| Per Base N Content | 0% across all positions. | High Ns indicate low signal-to-noise during sequencing. |
Experimental Protocol: Initial QC Workflow for ChIP-seq FASTQ Files
Objective: Assess the quality of raw sequencing reads prior to alignment. Materials: Raw paired-end or single-end FASTQ files from a transcription factor ChIP-seq experiment. Software: FastQC (v0.12.0+), MultiQC (v1.15+).
- Installation: Install via Conda:
conda create -n qc fastqc multiqc -c bioconda -c conda-forge - Run FastQC: Analyze all FASTQ files:
fastqc *.fastq.gz -o ./fastqc_results -t [number_of_threads] - Aggregate Reports: Generate a consolidated HTML report:
multiqc ./fastqc_results -o ./multiqc_report - Interpretation: Open the
multiqc_report.html. Focus on "Per base sequence quality," "Adapter Content," and "Sequence Duplication Levels." Use this to inform trimming parameters.
Diagram Title: FASTQ Quality Control and Trimming Workflow
The Scientist's Toolkit: Research Reagent Solutions for TF ChIP-seq
Table 3: Essential Reagents and Kits for TF ChIP-seq Library Prep
| Item | Function in Workflow | Example Vendor/Product |
|---|---|---|
| Specific Antibody | Immunoprecipitates the target transcription factor-DNA complex. Critical for success. | CST, Abcam, Diagenode; validated for ChIP. |
| Magnetic Protein A/G Beads | Captures antibody-bound complexes for washing and elution. | Thermo Fisher Dynabeads, Millipore Magna ChIP beads. |
| Chromatin Shearing Reagents | Enzymatic or sonication kits to fragment crosslinked chromatin to 150-500 bp. | Covaris sonication system, Diagenode Bioruptor, or enzymatic shearing kits. |
| Library Preparation Kit | Converts immunoprecipitated DNA into sequencing-ready libraries (end-repair, A-tailing, adapter ligation, PCR). | Illumina TruSeq ChIP Library Prep Kit, NEB Next Ultra II DNA Library Prep Kit. |
| Size Selection Beads | SPRI/AMPure beads to select library fragments of the correct size, removing primers and adapter dimers. | Beckman Coulter AMPure XP, KAPA Pure Beads. |
| High-Sensitivity DNA Assay | Quantifies final library concentration and assesses fragment size distribution prior to sequencing. | Agilent Bioanalyzer/TapeStation (HS DNA kit), Qubit dsDNA HS Assay. |
Pre-processing: Trimming and Filtering
Based on QC results, raw reads often require cleaning before alignment.
Experimental Protocol: Adapter Trimming and Quality Filtering with Trimmomatic
Objective: Remove adapter sequences and low-quality bases. Software: Trimmomatic (v0.39+).
- Command for Paired-End Reads:
- Parameter Explanation:
ILLUMINACLIP: Removes adapter sequences (specify adapter FASTA file).LEADING/TRAILING: Cut low-quality bases from start/end.SLIDINGWINDOW: Scans read with a 4-base window, cutting when average Q < 15.MINLEN: Discards reads shorter than 36 bp post-trimming.
After trimming, re-run FastQC to confirm improved metrics before proceeding to genome alignment in the ChIP-seq workflow.
Diagram Title: FASTQ Read Trimming Process
In transcription factor (TF) research using ChIP-seq, the alignment of sequencing reads to a reference genome is a critical computational step. This process translates short nucleotide sequences into genomic coordinates, enabling the identification of protein-DNA interaction sites. The accuracy, speed, and sensitivity of alignment directly impact downstream analyses, including peak calling and motif discovery, which are foundational for understanding gene regulation in development, disease, and drug discovery.
Core Principles of Read Alignment
Alignment involves mapping short reads (typically 50-300 bp) from a high-throughput sequencer to their most likely location in a large reference genome (e.g., human GRCh38). The central challenges include managing the vast search space, accounting for sequencing errors, and identifying genomic variations or true binding events. Key considerations are:
- Spliced vs. Unspliced Alignment: For ChIP-seq of transcription factors, unspliced alignment is standard, as TFs bind to genomic DNA, not spliced mRNA.
- Handling Multi-mapping Reads: Reads originating from repetitive genomic regions can map to multiple locations, requiring specialized strategies to avoid false positives.
- Accuracy Metrics: Mapping quality (MAPQ) scores assess alignment confidence, crucial for filtering in sensitive TF binding analyses.
Best Practices for ChIP-seq Read Alignment
- Quality Control Pre-Alignment: Use FastQC to assess read quality. Trimming adapters and low-quality bases with tools like Trimmomatic or Cutadapt is essential.
- Reference Genome Selection: Use the most current, primary assembly from a trusted source (e.g., GENCODE, Ensembl). Include decoy sequences to improve mapping of non-human reads and contaminants.
- Alignment Parameter Tuning: For TF ChIP-seq, allow for short gaps (indels) but typically disable long, splice-aware alignment. Set the
--no-spliced-alignmentflag in STAR or similar parameters in other aligners. - Post-Alignment Processing: Sort and index BAM files. Filter to remove duplicate reads (potential PCR artifacts) using tools like Picard MarkDuplicates, and exclude reads mapping to blacklisted regions (e.g., ENCODE Blacklist).
- Multi-mapping Read Handling: For broad peak factors or those binding repetitive elements, consider using alignment tools that retain multi-mappers or employing specialized peak callers that can utilize this information.
Quantitative Comparison of Leading Alignment Tools
The performance of aligners varies based on accuracy, speed, and memory usage. The following table summarizes key metrics based on recent benchmarking studies for human genomic data.
Table 1: Comparison of Common Read Aligners for ChIP-seq
| Tool | Algorithm Type | Speed (Relative) | Memory Usage | Best For ChIP-seq? | Key Consideration for TF Studies |
|---|---|---|---|---|---|
| Bowtie2 | FM-index, BWT | Moderate | Low | Excellent | Default settings well-suited for short-read (<100bp) TF ChIP-seq. |
| BWA-MEM | FM-index, BWT | Moderate | Low | Excellent | Robust for longer reads (70-300bp); good balance of speed and accuracy. |
| STAR | Spliced Alignment | Fast (in mapping mode) | High | Good (with flags) | Requires --alignIntronMax 1 to disable splicing for TF ChIP-seq. Very fast. |
| minimap2 | Minimizer-based | Very Fast | Low | Good | Efficient for long reads but also highly performant for short-read mapping. |
| Subread/Subjunc | Seed-and-vote | Fast | Moderate | Good | Designed for RNA-seq but alignment mode (subread-align) is accurate for DNA. |
Detailed Experimental Protocol: Alignment of TF ChIP-seq Reads
Protocol: From Raw FASTQ to Processed BAM for Transcription Factor ChIP-seq
I. Prerequisite Software & Data
- FastQC, Trimmomatic, chosen aligner (e.g., Bowtie2), SAMtools, Picard.
- Raw paired-end or single-end FASTQ files.
- Reference genome FASTA file and corresponding pre-built aligner index.
II. Step-by-Step Methodology
Quality Assessment:
fastqc sample_R1.fastq.gz sample_R2.fastq.gz -o ./fastqc_report/Adapter Trimming & Quality Filtering:
java -jar trimmomatic.jar PE -phred33 \sample_R1.fastq.gz sample_R2.fastq.gz \sample_R1_trimmed_paired.fq.gz sample_R1_trimmed_unpaired.fq.gz \sample_R2_trimmed_paired.fq.gz sample_R2_trimmed_unpaired.fq.gz \ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10 LEADING:3 TRAILING:3 \SLIDINGWINDOW:4:15 MINLEN:36Read Alignment (Bowtie2 Example):
bowtie2 -p 8 -x /path/to/genome_index \-1 sample_R1_trimmed_paired.fq.gz -2 sample_R2_trimmed_unpaired.fq.gz \--no-mixed --no-discordant --maxins 1000 \-S sample_aligned.samSAM to BAM Conversion & Sorting:
samtools view -@ 7 -bS sample_aligned.sam | \samtools sort -@ 7 -o sample_sorted.bamDuplicate Marking:
java -jar picard.jar MarkDuplicates \I=sample_sorted.bam \O=sample_marked.bam \M=marked_dup_metrics.txt \REMOVE_DUPLICATES=falseIndexing and Filtering (Optional):
samtools index sample_marked.bamsamtools view -@ 7 -q 10 -b sample_marked.bam > sample_filtered.bam
Visualizing the Alignment Workflow in ChIP-seq Analysis
ChIP-seq Read Alignment & Processing Workflow
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 2: Key Reagents and Materials for ChIP-seq Library Preparation and Validation
| Item | Function in TF ChIP-seq Workflow |
|---|---|
| Specific Antibody | Immunoprecipitates the target transcription factor-DNA complex. Must be validated for ChIP. |
| Protein A/G Magnetic Beads | Binds antibody-bound complexes for separation and washing. |
| Crosslinking Agent (Formaldehyde) | Fixes protein-DNA interactions in living cells prior to lysis. |
| Chromatin Shearing Reagents | Enzymatic (MNase) or sonication (Covaris) kits to fragment chromatin to 200-500 bp. |
| ChIP-seq Library Prep Kit | Contains enzymes and buffers for end repair, A-tailing, adapter ligation, and PCR amplification of immunoprecipitated DNA. |
| Size Selection Beads (SPRI) | Magnetic beads for clean-up and selection of appropriately sized DNA fragments post-library prep. |
| qPCR Primers | Validated primers for positive/negative genomic control regions to assess ChIP enrichment prior to sequencing. |
| High-Sensitivity DNA Assay Kit | Fluorometric quantification of low-concentration DNA libraries (e.g., Qubit). |
Within the broader thesis outlining a robust ChIP-seq workflow for transcription factor (TF) research, the step following alignment is critical: the Initial Quality Assessment (IQA). This phase, centered on mapping statistics and visual validation in the Integrative Genomics Viewer (IGV), determines if the data possesses the fundamental integrity required for downstream peak calling and motif analysis. A failure at this juncture can lead to erroneous biological conclusions regarding TF binding sites.
Quantitative Mapping Statistics: The First Indicator
Post-alignment files (typically BAM format) contain quantitative metrics that offer the first objective snapshot of experiment quality. Key statistics must be calculated and compared against field-established benchmarks. The following table summarizes these core metrics, their optimal ranges for TF ChIP-seq, and their biological interpretation.
Table 1: Core Mapping Statistics for TF ChIP-seq Quality Assessment
| Metric | Description | Optimal Range (TF ChIP-seq) | Interpretation & Implications |
|---|---|---|---|
| Total Reads | Total number of sequenced reads. | 20-50 million (for mammalian genomes) | Defines sequencing depth. Insufficient depth reduces peak detection sensitivity. |
| Aligned Reads (%) | Percentage of reads mapped to the reference genome. | >90% (varies by genome quality) | Low percentages indicate poor sample quality or contamination. |
| Uniquely Mapped Reads (%) | Percentage of reads mapped to a single genomic locus. | >70-80% | High multi-mapping reads can confound peak calling, especially for repeat-associated TFs. |
| Duplicate Rate (%) | Percentage of PCR or optical duplicates. | <20-30% (Post-deduplication) | High rates indicate over-amplification, reducing effective library complexity and statistical power. |
| Fraction of Reads in Peaks (FRiP) | Proportion of reads falling within called peak regions. | 1-5% (TF-specific; >1% is often acceptable) | Primary indicator of signal-to-noise. A low FRiP suggests poor enrichment or failed immunoprecipitation. |
| Cross-Correlation (NSC/ RSC) | Measures fragment length distribution and signal shift. | NSC > 1.05, RSC > 0.8 (ideally >1) | QC metric from ENCODE. Low scores indicate poor signal or background noise. |
Detailed Protocol: Generating Key Statistics
Protocol 1: Calculating Mapping and Duplicate Metrics using SAMtools and Picard
- Prerequisites: Installed SAMtools and Picard Toolkit. Sorted BAM file (
sample.sorted.bam). Calculate Alignment Statistics:
This outputs counts for total, primary, duplicate, mapped, and properly paired reads.
Mark Duplicates:
Index the BAM File:
Protocol 2: Calculating FRiP Score using BEDTools and Peak Caller Output
- Prerequisites: Installed BEDTools. Deduplicated BAM file and a BED file of called peaks (
sample_peaks.bed). Count Reads in Peaks:
Extract the total read count from
sample.flagstat.txt(from Protocol 1).- Calculate FRiP:
Visual Assessment in IGV: A Critical Qualitative Step
Quantitative metrics must be complemented by visual inspection in IGV to assess signal distribution, noise, and artifact presence.
Workflow for IGV Visualization:
- Load Data: Load the BAM alignment file (and its index,
.bai). Load a matched input/control BAM file for comparison. - Navigate to Positive and Negative Control Loci:
- Positive Control: Navigate to known, strong binding sites for the TF (e.g.,
MYCat promoter ofCDKN1A). Expect a dense, concentrated pileup of reads in the ChIP sample, minimal in the input. - Negative Control: Navigate to gene deserts or regions like the
GAPDHcoding sequence (lacking TF binding). Expect low, uniform read coverage in both ChIP and input.
- Positive Control: Navigate to known, strong binding sites for the TF (e.g.,
- Assess "Peakiness": The ChIP track should show sharp, localized enrichments ("peaks") against a low, flat background. A "puffy" or uniformly elevated signal indicates high background noise.
- Check for Artifacts: Look for anomalous, ultra-high coverage spikes (PCR artifacts) or repetitive patterns. Use IGV's "View as Paired" and "Show Splice Junctions" to inspect alignment integrity.
Title: IGV and Stats Quality Assessment Decision Workflow
The Scientist's Toolkit: Essential Reagent Solutions
Table 2: Key Research Reagents & Tools for ChIP-seq IQA
| Item | Function in IQA | Example/Note |
|---|---|---|
| High-Fidelity DNA Polymerase | Library amplification with minimal bias and error. Critical for maintaining library complexity and low duplicate rates. | KAPA HiFi, Q5 High-Fidelity. |
| Size Selection Beads | Precise isolation of adapter-ligated DNA fragments (~200-500 bp). Defines the insert size distribution visible in IGV. | SPRIselect (Beckman Coulter), AMPure XP. |
| Quantitative PCR (qPCR) Assay | Pre-sequencing validation using positive/negative control genomic loci. Predicts FRiP and confirms enrichment. | Primers for known binding sites vs. non-bound regions. |
| Phusion or Pfu Polymerase | For re-amplification of libraries if yield is low, though use cautiously to avoid exacerbating duplicates. | |
| Bioanalyzer/TapeStation | Quality control of final library fragment size distribution before sequencing. | Agilent Technologies. |
| IGV Software | Open-source visualization tool for interactive exploration of aligned read data against the reference genome. | Broad Institute. Essential for qualitative assessment. |
| SAMtools/Picard Suite | Command-line utilities for processing, sorting, indexing, and generating metrics from alignment files. | Essential for generating Table 1 statistics. |
Step-by-Step Computational Pipeline for TF Binding Site Detection
This guide details the critical pre-processing and filtering steps for ChIP-seq data analysis, a foundational component of a thesis on transcription factor (TF) research. Following sequencing, raw reads (FASTQ files) must be rigorously quality-controlled to eliminate technical artifacts and low-confidence data, ensuring subsequent peak calling and motif analysis accurately reflect true TF-DNA interactions. This stage directly impacts the validity of conclusions regarding TF binding sites, regulatory networks, and potential therapeutic targets in drug development.
Core Concepts and Quantitative Benchmarks
Defining Duplicates and Low-Quality Reads
- PCR Duplicates: Artifactual reads originating from PCR amplification during library preparation, appearing with identical start and end coordinates. They skew binding signal quantification.
- Optical Duplicates: A subset arising from clusters incorrectly identified as separate during sequencing imaging.
- Low-Quality Reads: Reads containing an excess of low-base-call-quality scores, adapter contamination, or an high proportion of ambiguous (N) bases.
Current Industry Standards and Thresholds
Table 1: Common Filtering Thresholds and Their Impact
| Metric | Typical Threshold | Rationale & Consequence of Not Filtering |
|---|---|---|
| PCR Duplicate Rate | < 20-30% for ChIP-seq | High rates indicate over-amplification, leading to spurious peak calls and inaccurate signal strength. |
| Adapter Content | > 5% triggers trimming | Adapter sequence contamination misaligns reads, causing loss of data and edge artifacts. |
| Low-Quality Bases (Q-score) | Q < 20-30 (Phred scale) | High probability of base-call error, leading to misalignment and false variant/SNP calls. |
| N-Content | > 5-10% of read length | Uncalled bases prevent unique alignment, reducing usable data. |
| Read Length | Post-trimming < 25-36 bp | Very short reads cannot be uniquely mapped to the reference genome. |
Detailed Methodologies and Protocols
Protocol for Adapter Trimming and Quality Filtering (using FastP)
This one-step protocol performs adapter trimming, quality pruning, and read filtering.
- Input: Paired-end or single-end FASTQ files.
- Software: fastP (v0.23.0+).
Command:
Parameters Explained:
--detect_adapter_for_pe: Auto-detects adapter sequences.--qualified_quality_phosphate 20: Bases with Q<20 are considered low-quality.--unqualified_percent_limit 40: Reads with >40% low-quality bases are discarded.--length_required 36: Reads shorter than 36bp after trimming are discarded.
- Output: Filtered FASTQ files and a comprehensive HTML quality report.
Protocol for Post-Alignment Duplicate Marking/Removal (using Picard)
Note: Duplicate marking is performed after alignment to the reference genome.
- Input: Coordinate-sorted BAM file from aligners like BWA or Bowtie2.
- Software: Picard Tools (v2.27+).
Command:
Parameters Explained:
REMOVE_DUPLICATES=false: Default behavior is to mark (flag) duplicates, not remove them, allowing for downstream analysis decisions.ASSUME_SORT_ORDER=coordinate: Input BAM must be coordinate-sorted.
- Output: BAM file with duplicates flagged (ready for removal by peak callers) and a metrics file detailing duplicate counts.
Visualization of Workflow Logic
ChIP-seq Pre-processing Logical Workflow
Title: ChIP-seq Read Pre-processing and Filtering Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools and Reagents for ChIP-seq Pre-processing
| Item / Solution | Function in Pre-processing Context |
|---|---|
| Illumina Sequencing Kits | Generate raw FASTQ data. Kit version dictates adapter sequences for trimming. |
| Standard Bioinformatic Suites | FastQC: Visualizes base quality, adapter content, Ns. MultiQC: Aggregates reports from multiple samples. |
| Trimming/Filtration Tools | fastP: All-in-one ultra-fast tool. Trimmomatic: Flexible, parameter-heavy trimmer. Cutadapt: Precise adapter removal. |
| Alignment Software | BWA-MEM / Bowtie2: Maps filtered reads to reference genome (hg38/mm10). Essential for coordinate-based duplicate marking. |
| Duplicate Marking Tools | Picard MarkDuplicates: Industry standard. sambamba markdup: Faster, parallelized alternative. |
| High-Performance Computing (HPC) or Cloud Resource | Required for storage and compute-intensive alignment and duplicate marking steps. |
| SAM/BAM Processing Tools | SAMtools: For sorting, indexing, and filtering aligned data post-marking. |
Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) is the cornerstone of in vivo transcription factor (TF) binding site identification. Within a comprehensive ChIP-seq workflow, peak calling—the computational detection of genomic regions enriched with aligned sequencing reads—is the critical step that transforms raw data into biological insights. The choice of algorithm directly impacts the sensitivity, specificity, and reproducibility of downstream analyses, including motif discovery, pathway enrichment, and drug target validation. This technical guide provides an in-depth comparison of three prominent peak callers: MACS2, HOMER, and the newer machine learning-based PeakDecks, framing their operation and performance within a robust TF research pipeline.
Core Algorithmic Methodologies
MACS2 (Model-based Analysis of ChIP-Seq 2)
MACS2 employs a dynamic Poisson distribution to model the genome-wide tag distribution, accounting for local biases.
- Remove Redundancy: Duplicate reads are filtered based on a user-defined threshold (default: one read per base pair).
- Shift Reads: Reads are shifted 5'->3' by d/2 to estimate the fragment size (d), centering the signal at the actual protein-DNA crosslinking point.
- Build Model: A sliding window (default: 100bp) scans the genome. For each window, a local λ is calculated from the read count in a larger surrounding region (default: 10,000bp) to model background noise.
- Peak Calling: A Poisson p-value is calculated for each window using the local λ. Regions significantly enriched over the background (default p-value < 1e-5) are called as peaks.
- Peak Merging & FDR Control: Overlapping peaks from forward and reverse strands are merged. A false discovery rate (FDR) is estimated by swapping the treatment and control samples.
HOMER (Hypergeometric Optimization of Motif EnRichment)
HOMER uses a peak-finding approach based on a fixed fragment size and a binomial/poisson background model, tightly integrated with de novo motif discovery.
- Define Tags: Reads are extended in the 3' direction by a predetermined fragment length (default: 75bp).
- Create Position Density Matrix: The genome is scanned to count tags at each position.
- Identify Enriched Regions: Contiguous regions where the tag density exceeds a given threshold (based on the local background) are identified.
- Statistical Scoring: Each region is scored using a binomial test (or Poisson) comparing tags in the region versus a background region (local genomic background or control input). Peaks are filtered based on a false discovery rate threshold.
- Integrated Motif Analysis: Called peaks are automatically passed to HOMER's motif finding algorithms to identify enriched DNA binding motifs.
PeakDecks
PeakDecks leverages a supervised machine learning framework, training a model to distinguish true peaks from background noise using multiple genomic features.
- Feature Extraction: For every candidate genomic window, a suite of features is extracted, including:
- Read count/summit strength
- Shape metrics (e.g., peak sharpness, skewness)
- Local mappability and GC content
- Signal-to-noise ratio relative to control.
- Model Prediction: A pre-trained gradient boosting model (e.g., XGBoost) evaluates the feature vector for each candidate window and outputs a probability score of being a true peak.
- Thresholding: Peaks are called by applying a threshold to the prediction score, which can be calibrated to achieve a desired precision-recall balance.
- Ensemble Approach: PeakDecks can integrate calls from multiple base callers (like MACS2 and HOMER) as features, potentially reconciling differences and improving consensus.
Quantitative Performance Comparison
Table 1: Algorithmic Characteristics & Requirements
| Feature | MACS2 | HOMER | PeakDecks |
|---|---|---|---|
| Core Model | Dynamic Poisson distribution | Binomial/Poisson test | Supervised Machine Learning (XGBoost) |
| Control Data | Recommended (for FDR) | Recommended (for background) | Highly Recommended |
| Primary Output | Narrow peaks (summits) | Broad regions | Narrow/Broad (adaptable) |
| Speed | Fast | Moderate | Slower (due to feature computation) |
| Ease of Use | Command-line, straightforward | Suite of tools, integrated workflow | Command-line, requires model/features |
| Key Strength | Robust default model, widely adopted | Integrated motif discovery & analysis | Potential for higher accuracy via multi-feature learning |
Table 2: Typical Performance Metrics on Benchmark TF ChIP-seq Datasets
| Metric | MACS2 | HOMER | PeakDecks |
|---|---|---|---|
| Sensitivity (Recall) | High | Moderate | Very High |
| Specificity (Precision) | High | High | Highest (on trained contexts) |
| Reproducibility (IDR)* | 0.94 - 0.98 | 0.92 - 0.96 | 0.96 - 0.99 |
| Summit Resolution | ~50-100bp | ~100-200bp | ~50-150bp |
| Memory Usage | Low | Moderate | High |
*IDR: Irreproducible Discovery Rate, lower is better.
Detailed Experimental Protocol for Comparative Validation
Objective: To benchmark MACS2, HOMER, and PeakDecks performance on a well-characterized transcription factor (e.g., CTCF) ChIP-seq dataset.
Materials: Public dataset (e.g., ENCODE: CTCF in GM12878 cells, accession ENCFF000VOX (ChIP) & ENCFF000VQE (Control)).
Software: Installed versions of macs2, homer (findPeaks), and PeakDecks.
Protocol:
Data Preprocessing:
- Download paired-end ChIP and Input control FASTQ files.
- Adapter trim with Trimmomatic:
java -jar trimmomatic.jar PE -phred33 R1.fastq.gz R2.fastq.gz ... - Align to reference genome (hg38) using BWA-MEM:
bwa mem -t 8 hg38.fa R1_trimmed.fq R2_trimmed.fq > aligned.sam - Convert to BAM, sort, and index using samtools.
- Filter duplicates using Picard Tools:
java -jar picard.jar MarkDuplicates I=input.bam O=deduplicated.bam M=metrics.txt
Peak Calling:
- MACS2:
macs2 callpeak -t ChIP_dedup.bam -c Input_dedup.bam -f BAMPE -g hs -n CTCF_MACS2 -B --call-summits - HOMER:
makeTagDirectory TagDir_ChIP/ ChIP_dedup.bamfollowed byfindPeaks TagDir_ChIP/ -style factor -o auto -i TagDir_Input/ - PeakDecks: First generate features, then predict:
peakdecks extract -c config.yamlthenpeakdecks predict -m model.pkl -f features.h5
- MACS2:
Benchmarking Analysis:
- Use published high-confidence CTCF binding sites from ENCODE as a gold standard.
- Calculate recall/sensitivity (fraction of gold standards recovered) and precision (fraction of called peaks overlapping gold standards) using BEDTools.
- Perform Irreproducible Discovery Rate (IDR) analysis using two biological replicates to assess consistency.
Visualization of Workflows and Logical Relationships
Diagram Title: ChIP-seq Analysis Workflow with Alternative Peak Callers
Diagram Title: Core Logic of Three Peak Calling Algorithms
The Scientist's Toolkit: Key Research Reagents & Solutions
Table 3: Essential Reagents and Materials for ChIP-seq & Validation
| Item | Function in TF ChIP-seq Workflow |
|---|---|
| Specific, High-Affinity Antibody | Immunoprecipitates the target transcription factor. Critical for signal-to-noise ratio. |
| Protein A/G Magnetic Beads | Efficient capture of antibody-protein-DNA complexes for washing and elution. |
| Formaldehyde | Crosslinks proteins to DNA to preserve in vivo binding interactions during cell lysis. |
| Glycine | Quenches formaldehyde crosslinking reaction. |
| Chromatin Shearing Reagents (Enzymatic or Sonication) | Fragments crosslinked chromatin to optimal size (200-600 bp) for sequencing. |
| DNA Clean-up & Size Selection Kits (e.g., SPRI beads) | Purify and select appropriately sized DNA fragments post-decrosslinking for library prep. |
| High-Fidelity PCR Master Mix | Amplifies the immunoprecipitated DNA library with minimal bias for sequencing. |
| Dual Indexing Adapters | Allows multiplexing of multiple samples in a single sequencing run. |
| qPCR Primers for Positive/Negative Genomic Loci | Validates ChIP enrichment efficiency prior to high-throughput sequencing. |
| Cell Line/Tissue with High TF Expression | Ensures sufficient starting material for robust signal detection. |
Within a comprehensive ChIP-seq data analysis workflow for transcription factor (TF) research, the parameter optimization of q-value thresholds, fold change (FC) cutoffs, and shift size is a critical step. This process directly influences the accuracy of peak calling, the biological relevance of identified binding sites, and the downstream interpretation of TF function in gene regulation. Improper settings can lead to high false discovery rates (FDR), loss of genuine binding events, or misalignment of paired-end reads, compromising the entire study. This guide provides an in-depth technical framework for optimizing these parameters, ensuring robust and reproducible results for drug development and mechanistic research.
Core Parameter Definitions and Impact
Table 1: Core Parameters in ChIP-seq Peak Calling
| Parameter | Definition | Biological/Statistical Impact | Typical Starting Range |
|---|---|---|---|
| q-value | The minimum false discovery rate (FDR) at which a peak is called. It is the adjusted p-value. | Controls the stringency of peak calling. Lower values reduce false positives but may increase false negatives. | 0.01 to 0.05 |
| Fold Change (FC) | The enrichment ratio of ChIP signal over background (control or input). | Determines the minimum enrichment required for a binding event. Higher values increase specificity but may miss weaker, biologically relevant sites. | 2 to 10 (linear scale) |
| Shift Size / Fragment Length | The estimated genomic distance between the two reads in a pair, or the shift applied to single-end reads to represent the sequenced fragment. | Critical for accurate peak positioning and resolution. Incorrect estimates smear or split peaks. | 100-300 bp |
Methodologies for Parameter Optimization
Empirical Optimization of q-value and Fold Change
Protocol: Cross-referencing with Biological Validation
- Iterative Peak Calling: Run your peak caller (e.g., MACS2) with a matrix of parameters: q-values (e.g., 0.001, 0.01, 0.05, 0.1) and fold-change thresholds (e.g., 2, 4, 8, 10).
- Assess Consistency: Compare the peak sets from replicates for each parameter combination using metrics like Irreproducible Discovery Rate (IDR).
- Biological Ground Truth: If available, intersect peaks from each condition with known binding motifs (from databases like JASPAR), conserved genomic regions, or previously validated binding sites from literature.
- Functional Enrichment: Perform Gene Ontology (GO) or pathway enrichment analysis on genes associated with each peak set. Optimal parameters often yield the most biologically plausible enrichment.
- Select Optimal Set: Choose the parameter pair that maximizes the balance between reproducibility (high IDR score), motif enrichment (lowest p-value for known TF motif), and functional coherence.
Table 2: Sample Parameter Optimization Results for a TF 'X'
| q-value | Fold Change | Peaks Called | % Peaks with Known Motif | IDR < 0.05 (Reproducibility) |
|---|---|---|---|---|
| 0.001 | 4 | 5,201 | 85% | 95% |
| 0.01 | 4 | 12,847 | 78% | 92% |
| 0.05 | 4 | 25,632 | 65% | 85% |
| 0.01 | 2 | 31,559 | 60% | 80% |
| 0.01 | 8 | 8,112 | 82% | 94% |
Experimental Determination of Shift Size/Fragment Length
Protocol: Wet-Lab and Computational Estimation
- Wet-Lab Estimation (Gold Standard):
- Run the ChIP-seq library on a Bioanalyzer or TapeStation.
- Measure the modal size of the fragment distribution in the library post-size-selection but prior to sequencing.
- This physical measurement provides the ground-truth shift/fragment length.
- Computational Estimation (MACS2):
- For paired-end data: The shift is inherently determined by the read alignment. Use
samtools statsto check insert size distribution. - For single-end data: Use the
macs2 predictdfunction on the aligned input/control sample. - Input:
macs2 predictd -i input.bam -g hs(for human). - Output: A model showing the peak of the fragment length distribution. Visually inspect the generated plot to confirm a clear bimodal pattern.
- For paired-end data: The shift is inherently determined by the read alignment. Use
Integrated Workflow for Parameter Setting
Diagram Title: ChIP-seq Parameter Optimization Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Tools for ChIP-seq Parameter Optimization
| Item | Function in Parameter Optimization |
|---|---|
| High-Sensitivity DNA Assay (e.g., Agilent Bioanalyzer HS DNA kit) | Precisely measures post-ChIP library fragment size distribution, providing the ground-truth for shift/fragment length parameter. |
| High-Fidelity PCR Master Mix (e.g., NEB Next Ultra II) | Ensures unbiased amplification during library prep, maintaining the original fragment length distribution critical for accurate shift estimation. |
| SPRIselect Beads (e.g., Beckman Coulter) | Enables precise size selection of libraries, which directly defines the fragment length range analyzed and impacts shift size. |
| Validated Positive Control Antibody (e.g., anti-RNA Pol II) | Provides a benchmark dataset with well-characterized peaks to test and calibrate q-value/FC thresholds for a new experiment. |
| Commercial Peak Caller Software/Suite (e.g., HOMER, Partek Flow) | Often include built-in diagnostic plots and optimization modules for shift size, q-value, and FC, streamlining the process. |
| Genomic DNA Spike-in Control (e.g., from D. melanogaster) | Allows for normalization and assessment of signal-to-noise, informing appropriate FC cutoff selection, especially for differential binding studies. |
Advanced Considerations: Differential Binding and Drug Treatment
In studies involving drug treatments or disease states, differential binding analysis adds complexity. The chosen q-value/FC thresholds for initial peak calling should be lenient enough to capture all potential sites (e.g., q=0.05), with stringent statistical thresholds applied subsequently during differential analysis (e.g., FDR < 0.1 & log₂FC > 1). The shift size, however, remains an experiment-level property and should be consistent across all samples in a cohort.
Diagram Title: Differential Binding Analysis Workflow
Systematic optimization of q-values, fold change, and shift size is non-negotiable for deriving biologically actionable insights from ChIP-seq data in transcription factor research. By integrating wet-lab measurements, computational diagnostics, and iterative validation against biological knowledge, researchers can establish a rigorous foundation for their analysis pipeline. This diligence ensures that subsequent conclusions regarding transcriptional mechanisms, disease-associated dysregulation, or drug-induced effects are built upon a reliable and accurate set of transcription factor binding events.
Within the comprehensive thesis on ChIP-seq data analysis workflows for transcription factor (TF) research, a critical bifurcation exists in peak calling and downstream interpretation. This divergence is fundamentally dictated by the nature of the protein of interest: sequence-specific transcription factors, which produce narrow, punctate peaks, and broad histone modifications, which generate expansive, diffuse enrichment domains. Accurately handling this distinction is not merely a technical detail but a core determinant for deriving biologically meaningful conclusions in gene regulation studies and subsequent drug discovery efforts.
Defining Characteristics and Biological Basis
The physical interaction patterns observed in ChIP-seq assays are direct readouts of protein-DNA binding dynamics.
Narrow Peaks (Transcription Factors): TFs bind to specific, short consensus sequences (e.g., E-box, AP-1 site) for relatively brief periods. This results in sharp, high-intensity enrichment signals typically spanning 50-500 bp. These peaks precisely mark transcription factor binding sites (TFBS) and are often located in promoters, enhancers, and insulators.
Broad Domains (Histone Marks): Histone modifications, such as H3K36me3 (transcription elongation) or H3K27me3 (polycomb repression), are deposited across large genomic regions encompassing entire gene bodies or broad regulatory landscapes. These marks produce wide, lower-amplitude enrichment regions that can span several kilobases to over 100 kb.
Quantitative Comparison of Peak Profiles
| Feature | Transcription Factor (Narrow) Peaks | Broad Histone Mark Domains |
|---|---|---|
| Typical Genomic Width | 50 - 500 base pairs | 5,000 - 100,000+ base pairs |
| Peak Shape | Sharp, punctate | Wide, plateau-like or rolling hills |
| Canonical Examples | p53, CTCF, NF-κB, ERα | H3K27me3, H3K36me3, H3K9me3 |
| Primary Biological Signal | Direct protein-DNA binding event | Chromatin state and epigenetic landscape |
| Optimal Peak Caller Examples | MACS2, HOMER, GEM | SICER2, BroadPeak, SEACR, RSEG |
| Typical Sequencing Depth | 20-40 million reads (high depth for sensitivity) | 30-60 million reads (depth for broad signal) |
| Key Analysis Metric | Peak summit precision, motif enrichment | Domain stability, enrichment breadth |
Experimental Protocols for Differential Analysis
Protocol 1: ChIP-seq for a Transcription Factor (e.g., p53)
1. Crosslinking & Cell Harvesting: Treat cells (e.g., MCF-7) with appropriate stimulus (e.g., Doxorubicin for p53 activation). Fix protein-DNA interactions with 1% formaldehyde for 10 min at room temperature. Quench with 125 mM glycine. 2. Sonication: Lyse cells and shear chromatin to an average fragment size of 150-500 bp using a focused ultrasonicator (e.g., Covaris S220). Verify size distribution on a 2% agarose gel. 3. Immunoprecipitation: Incubate sheared chromatin with 2-5 µg of validated, high-specificity anti-p53 antibody (e.g., DO-1) bound to magnetic Protein A/G beads overnight at 4°C. Include an isotype control IgG sample. 4. Washing & Elution: Wash beads with low-salt, high-salt, LiCl, and TE buffers. Reverse crosslinks by incubating with elution buffer (1% SDS, 0.1M NaHCO3) and 200 mM NaCl at 65°C overnight. 5. Library Preparation & Sequencing: Purify DNA, end-repair, A-tail, and ligate sequencing adapters. Amplify with 12-18 PCR cycles. Perform 50-75 bp single-end sequencing on an Illumina platform to a depth of 25-40 million mapped reads.
Protocol 2: ChIP-seq for a Broad Histone Mark (e.g., H3K27me3)
1. Crosslinking & Harvesting: Fix cells as above. For some histone marks, native ChIP (without crosslinking) can be performed. 2. Sonication: Shear chromatin to a slightly larger average size (200-700 bp) to help capture broad domains. 3. Immunoprecipitation: Use 2-5 µg of highly specific antibody (e.g., C36B11 for H3K27me3). Due to lower signal-to-noise, rigorous controls are essential. 4. Washing & Elution: Use standard IP wash buffers. Elute as above. 5. Library Preparation & Sequencing: Construct libraries as above. Sequence to a higher depth (40-60 million reads) to ensure sufficient coverage across broad, low-amplitude regions. Paired-end sequencing (e.g., 75 bp PE) is beneficial.
Computational Analysis Workflow
Figure 1: ChIP-seq analysis workflow bifurcation.
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function in ChIP-seq | Key Considerations |
|---|---|---|
| Formaldehyde (1%) | Reversible protein-DNA crosslinking. | Over-fixing increases background; optimize incubation time. |
| High-Specificity Primary Antibody | Immunoprecipitation of target protein or histone mark. | Validate for ChIP (ChIP-grade). High titer and specificity are critical for signal-to-noise. |
| Magnetic Protein A/G Beads | Capture antibody-target complexes. | Superior recovery and lower background vs. agarose beads. |
| Covaris S220 Ultrasonicator | Shearing chromatin to optimal fragment size. | Provides consistent, tunable shearing; minimizes over-shearing. |
| PCR-Free or Low-Cycle Library Prep Kit | Amplification of immunoprecipitated DNA for sequencing. | Minimizes PCR duplicates and bias. Essential for quantitative analysis. |
| SPRI Beads (e.g., AMPure XP) | Size selection and cleanup of DNA fragments. | Reproducible alternative to gel extraction. |
| High-Fidelity DNA Polymerase | Amplification of ChIP libraries. | Reduces errors during PCR steps of library prep. |
| Validated Control Antibodies | Positive control (e.g., H3K4me3) and negative control (IgG). | Essential for assessing experiment success and background subtraction. |
Signaling Pathway Context for TF Binding
Figure 2: TF binding in cellular signaling context.
Downstream Analytical Considerations
Beyond peak calling, subsequent analyses diverge. For narrow TF peaks, the focus is on motif discovery to identify the bound sequence and nearest gene annotation for linking TFBS to potential target genes. For broad marks, analysis shifts to domain segmentation of the genome into distinct chromatin states and gene body enrichment assessment (e.g., H3K36me3 across transcribed regions). Both data types converge in integrative analysis, where TF binding sites are overlaid with chromatin states to elucidate enhancer-promoter interactions and regulatory networks, a cornerstone for identifying therapeutic targets in disease.
The dichotomy between narrow TF peaks and broad histone marks necessitates a tailored, biologically informed approach at every stage of the ChIP-seq workflow, from experimental design through computational analysis. Recognizing and respecting this distinction is fundamental within the larger thesis of a robust ChIP-seq pipeline, ensuring accurate interpretation of gene regulatory mechanisms and providing a solid foundation for research in molecular biology and targeted drug development.
Within the comprehensive ChIP-seq data analysis workflow for transcription factor (TF) research, the critical step following peak calling is peak annotation. This process bridges the gap between identifying genomic regions bound by a TF (the peaks) and interpreting their potential biological function by associating them with nearby or overlapping genes and genomic features.
Core Concepts and Quantitative Context
The primary goal is to determine the probable target genes regulated by the TF of interest. This is inferred based on the genomic proximity of a binding peak to a gene's transcriptional start site (TSS) or regulatory elements. The distribution of peaks across different genomic features is rarely uniform.
Table 1: Typical Distribution of ChIP-seq Peaks Across Genomic Features
| Genomic Feature | Approximate Percentage of Peaks | Functional Implication |
|---|---|---|
| Promoter (≤ 1kb from TSS) | 20-40% | Direct transcriptional regulation via core promoter machinery. |
| 5' UTR / Exonic | 2-8% | Potential involvement in transcriptional elongation or RNA processing. |
| Intronic | 20-35% | Often contains enhancers or silencers; cell-type specific regulation. |
| Distal Intergenic | 30-50% | Likely candidate enhancer or repressor regions; requires long-range interaction analysis. |
| 3' UTR | 1-5% | Potential role in mRNA stability or translation. |
Table 2: Common Genomic Annotation Databases & Resources
| Resource Name | Type | Key Use in Peak Annotation |
|---|---|---|
| ENSEMBL | Genome Database | Provides comprehensive gene models, TSS coordinates, and biotype information. |
| UCSC RefSeq | Genome Database | Curated gene annotations; often used for standard genomic coordinates. |
| GENCODE | Genome Annotation | High-quality manual annotation, especially for non-coding genes and complex loci. |
| FANTOM/CAGE | TSS Atlas | Defines precise, cell-type specific TSS locations for accurate promoter linkage. |
Detailed Experimental Protocol: Proximity-Based Peak-to-Gene Annotation
This protocol uses bioinformatics tools to assign peaks to genes based on nearest TSS distance.
Materials & Software:
- BED file of called peaks from MACS2 or similar caller.
- Reference genome annotation file (GTF/GFF3 format) from ENSEMBL, RefSeq, or GENCODE.
- Computer with UNIX/Linux environment and sufficient RAM (≥16 GB recommended).
- Bioinformatics tools: BEDTools, R/Bioconductor with packages like
ChIPseeker,ChIPpeakAnno, orHOMER.
Procedure:
Step 1: Data Preparation
- Ensure peak file is in BED format (chromosome, start, end, name, score, strand...).
- Download the appropriate GTF annotation file for your reference genome assembly (e.g., GRCh38.p13, mm10).
- In a terminal, use
grepto extract only "gene" or "transcript" features from the GTF to simplify the annotation:
Step 2: Annotate Peaks Using BEDTools (Command-Line Method)
- Use
bedtools closestto find the nearest gene TSS for each peak. First, create a BED file of TSS coordinates from the GTF.
- The
-D refoption reports the distance of the peak to the TSS, with negative values indicating upstream.
Step 3: Annotate Peaks Using R/Bioconductor (ChIPseeker)
- In R, load the peak file and annotate using the
annotatePeakfunction, which provides rich genomic context.
ChIPseekercategorizes peaks into Promoter, 5' UTR, 3' UTR, Exon, Intron, Downstream, and Distal Intergenic regions.
Step 4: Functional Enrichment Analysis
- Use the list of annotated genes (e.g., those with peaks in their promoter) as input for Gene Ontology (GO) or pathway analysis (KEGG, Reactome) using packages like
clusterProfiler.
Visualizing the Peak Annotation Workflow
ChIP-seq Peak Annotation and Analysis Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents & Kits for Experimental Validation of Annotated Peaks
| Item Name | Function in Downstream Validation | Example Vendor/Cat. No. (Illustrative) |
|---|---|---|
| Chromatin Immunoprecipitation (ChIP) Kit | Validates TF binding at specific annotated loci identified in silico. Essential for confirming peak authenticity. | MilliporeSigma (17-295), Cell Signaling (#9005) |
| qPCR Probes/Primers | Designed for sequences within annotated peaks and control regions. Quantifies enrichment from validation ChIP. | Thermo Fisher Scientific (TaqMan Assays), IDT (PrimeTime qPCR Probes) |
| Dual-Luciferase Reporter Assay System | Tests the enhancer/promoter activity of genomic regions identified as peaks, cloned upstream of a minimal promoter. | Promega (E1910) |
| CRISPR/dCas9 Activation or Interference Systems | Functionally links annotated distal peaks to target genes by perturbing the peak region and measuring gene expression changes. | Santa Cruz Biotechnology (sc-400206), Takara Bio (632607) |
| High-Fidelity DNA Polymerase | Amplifies predicted peak regions for cloning into reporter vectors or for generating probes. | NEB (M0491S), Kapa Biosystems (KK2101) |
| Gel Extraction & Plasmid Purification Kits | Isolates specific DNA fragments (peak regions) for downstream cloning and reporter assays. | Qiagen (28704, 27104) |
Advanced Considerations: Beyond Simple Proximity
Proximity-based annotation has limitations, especially for distal intergenic peaks that may regulate genes via long-range chromatin loops. Integrating additional data is crucial for a robust thesis.
- Chromatin Conformation Data (Hi-C, ChIA-PET): Provides physical interaction maps to link distal enhancers (peaks) to target promoters.
- Chromatin State Segmentation (from histone marks): Helps classify peaks into active enhancers, poised enhancers, or repressed regions using tools like ChromHMM or Segway.
- Co-binding with other TFs or Co-activators (p300, Mediator): Supports the functional importance of an annotated peak.
Linking Distal Peaks to Genes via Chromatin Looping
This integrated approach to peak annotation—combining proximity, chromatin states, and interaction data—transforms a simple list of genomic coordinates into a functional map of a transcription factor's regulatory network, forming a cornerstone for subsequent mechanistic studies and therapeutic target identification in drug development.
Motif discovery is a critical, downstream analytical step in a comprehensive ChIP-seq data analysis workflow for transcription factor (TF) research. Following peak calling—which identifies genomic regions enriched for TF binding—motif analysis interrogates these regions to decipher the sequence code that directs TF occupancy. This process validates the ChIP experiment by confirming that the immunoprecipitated factor binds its expected sequence and can reveal novel, co-binding partners. Within drug development, understanding these precise recognition rules is fundamental for identifying dysregulated transcriptional programs in disease and for designing therapeutics that modulate TF activity.
Core Concepts:De Novovs. Known Motif Discovery
- De Novo Motif Discovery: The ab initio identification of overrepresented sequence patterns within a set of genomic regions (e.g., ChIP-seq peaks) without prior sequence models. It answers: "What sequence motifs are enriched in my peaks?"
- Known Motif Scanning (or Matching): The comparison of identified peaks against databases of previously characterized TF binding motifs. It answers: "Does my dataset contain binding sites for known factor X or its relatives?"
Table 1: Comparison of De Novo and Known Motif Discovery Approaches
| Aspect | De Novo Discovery | Known Motif Scanning |
|---|---|---|
| Primary Goal | Identify novel, unknown sequence motifs. | Annotate peaks with potential binding factors. |
| Input | FASTA sequences from ChIP-seq peaks. | FASTA sequences + a database of Position Weight Matrices (PWMs). |
| Key Algorithms | MEME, DREME, HOMER. | FIMO, AME, HOMER (scanning module). |
| Output | One or more novel motifs represented as PWMs. | A list of known motifs significantly enriched in the input sequences. |
| Main Challenge | Computational intensity; distinguishing true signals from background. | Managing false positives from motif similarity; database completeness. |
Detailed Experimental & Computational Protocols
Protocol A:De NovoMotif Discovery with HOMER
Objective: To find the most significantly enriched DNA sequence motifs in a set of ChIP-seq peak regions.
Materials & Input:
- A BED file of high-confidence ChIP-seq peaks (
peaks.bed). - Reference genome FASTA file (e.g.,
hg38.fa). - HOMER software suite installed.
Procedure:
- Convert Peaks to Sequences:
- Execute De Novo Discovery:
- Interpretation: Results are in
./motif_output/. The filehomerResults.htmlshows ranked motifs. The primary output is a set of PWMs (e.g.,motif1.motif,motif2.motif).
Protocol B: Known Motif Enrichment Analysis with MEME Suite (AME)
Objective: To statistically test if known motifs from a database are enriched in ChIP-seq peaks compared to a background set.
Materials & Input:
- FASTA file of peak sequences (
peaks.fa). - FASTA file of matched background sequences (e.g., genomic regions with similar GC content;
background.fa). - A database of known PWMs (e.g., JASPAR
JASPAR2024_CORE_vertebrates_non-redundant.memeformat).
Procedure:
- Prepare Background: Generate control sequences using
shuffleSequences.pl(HOMER) orfasta-shuffle-letters(MEME). - Run AME (Analysis of Motif Enrichment):
- Interpretation: The output
ame.htmlprovides an E-value (significance) and p-value for each tested motif. A significant result indicates the known motif is overrepresented in the peak set.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Resources for Motif Discovery in ChIP-seq Analysis
| Item | Function/Description | Example Tools/Databases |
|---|---|---|
| ChIP-seq Peak Caller | Identifies genomic regions of significant TF binding from aligned sequencing data. | MACS3, HOMER findPeaks, SPP. |
| Sequence Extraction Tool | Converts genomic coordinates (BED files) to nucleotide sequences (FASTA). | BEDTools getfasta, HOMER annotatePeaks.pl. |
| De Novo Motif Finder | Discovers novel, enriched sequence patterns without prior information. | MEME, DREME, HOMER findMotifsGenome.pl. |
| Motif Scanning Tool | Searches sequences for matches to a given PWM. | FIMO, HOMER scanMotifGenomeWide.pl. |
| Motif Enrichment Tool | Tests statistical enrichment of known motifs against background. | AME, HOMER findMotifsGenome.pl (known). |
| PWM Database | Curated collection of transcription factor binding motifs. | JASPAR, CIS-BP, HOCOMOCO. |
| Motif Comparison Tool | Quantifies similarity between motifs, aiding in identification. | TOMTOM, STAMP. |
| Genome Browser | Visualizes motif locations relative to peaks and genomic annotations. | IGV, UCSC Genome Browser. |
Table 3: Example Output from a Combined Motif Discovery Analysis
| Motif Rank | Motif Logo | E-value / p-value | Best Match in JASPAR (TOMTOM) | Putative TF |
|---|---|---|---|---|
| 1 | ![Motif1] | 1.2e-25 (de novo) | MA0144.2 (p=3.1e-07) | NRF1 |
| 2 | ![Motif2] | 5.8e-12 (de novo) | MA0036.1 (p=1.4e-03) | MYC |
| 3 | - | 2.3e-30 (AME) | MA0516.1 | TP53 |
| 4 | - | 7.1e-18 (AME) | MA0079.3 | SP1 |
Note: E-value/p-value thresholds for significance are typically < 0.05 or < 1e-5, depending on the tool and multiple-testing correction applied.
Visualization of Workflows
ChIP-seq to Motif Discovery Workflow
Choosing a Motif Discovery Strategy
This whitepaper provides an in-depth technical guide for integrating ChIP-seq and RNA-seq data to establish causal links between transcription factor (TF) binding and transcriptional outcomes. This integrative analysis is a critical component of a comprehensive ChIP-seq data analysis workflow for transcription factor research, enabling researchers and drug development professionals to move beyond correlation and toward mechanistic understanding.
Foundational Concepts and Quantitative Data
The core premise is that TF binding, as measured by ChIP-seq, directly or indirectly regulates the expression of target genes, measured by RNA-seq. Key quantitative relationships and metrics are summarized below.
Table 1: Core Metrics in Integrative TF Binding-Gene Expression Analysis
| Metric | Typical Data Source | Purpose/Interpretation | Common Tools for Calculation |
|---|---|---|---|
| Peak-Gene Linkage | ChIP-seq | Defines putative target genes for a TF based on genomic proximity or chromatin interaction. | bedtools closest, HOMER, GREAT |
| Differential Binding (DB) | ChIP-seq (multiple conditions) | Identifies genomic regions with significant changes in TF occupancy between conditions. | DESeq2, edgeR, MACS2/diffBind |
| Differential Expression (DE) | RNA-seq (multiple conditions) | Identifies genes with significant changes in expression level between conditions. | DESeq2, edgeR, limma-voom |
| Expression-Binding Correlation | Integrated ChIP-seq & RNA-seq | Measures statistical association between TF binding strength (e.g., read count) and target gene expression level across samples. | Custom R/Python scripts |
| Overlap Significance | Integrated DB & DE results | Determines if the overlap between differentially bound genes and differentially expressed genes is greater than expected by chance (e.g., Fisher's Exact Test). | R (stats package), online enrichment tools |
Table 2: Common Genomic Proximity Criteria for Peak-Gene Assignment
| Assignment Rule | Typical Distance | Advantage | Limitation |
|---|---|---|---|
| Nearest TSS | Variable | Simple, unambiguous. | May assign peaks to unrelated distal genes. |
| Fixed Window around TSS | e.g., ±5 kb to ±50 kb | Captures common promoter-proximal regulation. | Misses long-range enhancers; includes many non-functional associations. |
| Within same TAD | ~100 kb - 1 Mb | Biologically informed by 3D chromatin architecture. | Requires Hi-C data which may not be available. |
Experimental Protocols
Protocol: Matched ChIP-seq and RNA-seq Sample Preparation
Objective: Generate high-quality, biologically paired datasets from the same cell population or tissue under identical conditions.
- Cell/Tissue Harvest: Split a homogenous cell population or pulverized tissue aliquot into two portions.
- Crosslinking (for ChIP-seq): Fix one portion with 1% formaldehyde for 8-12 minutes at room temperature. Quench with 125mM glycine.
- Cell Lysis & Chromatin Shearing (for ChIP-seq): Lyse cells and sonicate chromatin to achieve fragments of 200-500 bp. Verify size by gel electrophoresis.
- Immunoprecipitation (for ChIP-seq): Incubate sheared chromatin with validated, high-specificity antibody against the target TF. Capture antibody-chromatin complexes with protein A/G beads. Wash stringently.
- Reverse Crosslinking & Purification (for ChIP-seq): Elute complexes, reverse crosslinks at 65°C, and purify DNA (ChIP-seq library input).
- *RNA Stabilization (for RNA-seq): Immediately lyse the second, non-crosslinked portion in TRIzol or a similar RNase-inhibiting buffer. Store at -80°C.
- RNA Extraction & DNase Treatment (for RNA-seq): Isolate total RNA, treat with DNase I to remove genomic DNA contamination.
- Library Preparation: Construct sequencing libraries for both ChIP-seq (from purified DNA) and RNA-seq (from purified RNA, typically poly-A selected or rRNA-depleted) using standard Illumina-compatible protocols.
- Sequencing: Sequence ChIP-seq libraries (typically 20-50 million single-end 50-75 bp reads) and RNA-seq libraries (typically 25-40 million paired-end 100-150 bp reads) on an Illumina platform.
Protocol: Integrative Bioinformatics Analysis Workflow
Objective: Process paired datasets to identify significant TF-bound genes whose expression changes.
- ChIP-seq Processing:
a. Alignment: Map reads to reference genome (e.g., hg38) using
BWAorBowtie2. b. Peak Calling: Identify significant regions of enrichment (peaks) usingMACS2. c. Differential Binding: If multiple conditions exist, usediffBind(utilizingDESeq2/edgeR) to call DB regions. - RNA-seq Processing:
a. Alignment/Quantification: Map reads and quantify gene-level counts using
STAR+featureCountsor a pseudo-aligner likeSalmon. b. Differential Expression: UseDESeq2oredgeRto identify DE genes between conditions. - Integration & Assignment:
a. Link Peaks to Genes: Assign ChIP-seq peaks to gene promoters (e.g., TSS ± 5kb) using
bedtools closestor regulatory domain tools like GREAT. b. Overlap Analysis: Perform statistical enrichment (Fisher's Exact Test) to test if genes near DB peaks are significantly enriched among DE genes. c. Visualization: Create scatter plots of binding signal vs. expression, or genomic browser tracks overlaying ChIP-seq and RNA-seq data.
Figure 1: Workflow for integrative ChIP-seq and RNA-seq analysis.
Figure 2: Pathway linking TF binding to gene expression changes.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Integrative TF Binding & Expression Studies
| Item | Function / Rationale | Example Product/Kit |
|---|---|---|
| High-Specificity TF Antibody (ChIP-grade) | Essential for specific immunoprecipitation of the TF-DNA complex in ChIP-seq. Validation for ChIP is critical. | Cell Signaling Technology ChIP-validated Abs, Abcam ChIP-seq grade Abs. |
| Magnetic Protein A/G Beads | Efficient capture and washing of antibody-bound chromatin complexes. | Dynabeads Protein A/G, Millipore Magna ChIP Protein A/G Beads. |
| Formaldehyde (Ultra Pure) | Reversible crosslinking agent to fix protein-DNA interactions in living cells/tissue. | Thermo Scientific Pierce 16% Formaldehyde (w/v), Methanol-free. |
| Chromatin Shearing System | Fragmentation of crosslinked chromatin to optimal size (200-500 bp) for resolution. | Covaris ultrasonicator, Bioruptor Pico (diagenode). |
| RNase Inhibitor & RNA Stabilization Reagent | Preserves RNA integrity during sample splitting for matched RNA-seq. | Invitrogen SUPERase•In, QIAGEN RNAlater. |
| Total RNA Isolation Kit | High-yield, high-purity RNA extraction, often with integrated DNase treatment. | Zymo Research Quick-RNA Miniprep Kit, Qiagen RNeasy Plus Kit. |
| Stranded RNA-seq Library Prep Kit | Converts purified RNA into sequencer-compatible libraries, preserving strand information. | Illumina Stranded mRNA Prep, NEBNext Ultra II Directional RNA. |
| ChIP-seq DNA Library Prep Kit | Prepares sequencing libraries from low-input, fragmented ChIP DNA. | NEBNext Ultra II DNA Library Prep, KAPA HyperPrep Kit. |
| Dual Indexing Primers (Unique Dual Indexes - UDIs) | Enables pooled sequencing of multiple libraries from both RNA-seq and ChIP-seq runs, reducing index hopping. | Illumina UDI Sets, IDT for Illumina UDI. |
Solving Common ChIP-seq Problems and Improving Signal-to-Noise Ratio
Within the comprehensive thesis of a ChIP-seq workflow for transcription factor (TF) research, the critical bottleneck is often data quality. Successful TF ChIP-seq hinges on achieving high specific signal (enrichment) over low non-specific noise (background). This guide diagnoses the root causes of poor signal—low enrichment and high background—and provides technical solutions to rectify them at each experimental and computational stage.
Quantifying the Problem: Key Metrics
Poor data quality is quantifiable through established metrics, summarized in Table 1.
Table 1: Key Metrics for Diagnosing ChIP-seq Data Quality
| Metric | Optimal Range (TF ChIP-seq) | Indicative of Low Enrichment | Indicative of High Background | Common Assessment Tool |
|---|---|---|---|---|
| FRiP (Fraction of Reads in Peaks) | 1-5%+ (TF-specific) | < 1% | N/A | peakcaller output (e.g., MACS2) |
| NSC (Normalized Strand Cross-correlation) | > 1.05 (≥1.1 ideal) | ≤ 1.05 | N/A | phantompeakqualtools |
| RSC (Relative Strand Cross-correlation) | > 0.8 (≥1 ideal) | < 0.8 | < 0.8 | phantompeakqualtools |
| Number of Peaks | Protocol/ TF-dependent | Drastically low count | Excessively high count | MACS2, SEACR |
| Peak-Shape Metrics | Sharp, narrow peaks | Broad, diffuse peaks | Broad, diffuse peaks | visualization (IGV) |
| Library Complexity (NRF, PBC1) | NRF > 0.9, PBC1 > 0.9 | Low values | Low values | preseq, picard tools |
Experimental Protocol: A Rigorous QC ChIP-seq Workflow
The following detailed protocol incorporates critical quality control steps to mitigate poor signal.
A. Cell Fixation & Lysis
- Materials: 1-2x10^7 cells per IP, 37% formaldehyde, 2.5M glycine, cold PBS, cell lysis buffer (10 mM Tris-HCl pH 8.0, 85 mM KCl, 0.5% NP-40, protease inhibitors).
- Method:
- Cross-link cells with 1% formaldehyde for 8-12 minutes at room temperature with gentle agitation. Over-fixation increases background.
- Quench with 125 mM glycine (final conc.) for 5 min.
- Wash 2x with cold PBS. Pellet cells and flash-freeze or proceed.
- Resuspend pellet in 1 mL cell lysis buffer, incubate on ice 15 min.
- Centrifuge (5,000g, 5 min, 4°C). Discard supernatant.
B. Chromatin Shearing & Pre-Clear
- Materials: Sonication buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS, protease inhibitors), Bioruptor or Covaris, protein A/G magnetic beads, dilution buffer (16.7 mM Tris-HCl pH 8.0, 167 mM NaCl, 1.2 mM EDTA, 1.1% Triton X-100, 0.01% SDS).
- Method:
- Resuspend nuclear pellet in 1 mL sonication buffer. Sonicate to achieve 100-500 bp fragments (optimize per cell type). Insufficient shearing causes high background.
- Centrifuge (20,000g, 15 min, 4°C). Transfer supernatant (chromatin) to new tube.
- Dilute chromatin 1:10 in dilution buffer. Take 1% as "Input" control.
- Pre-clear with 20 μL protein A/G beads (per IP) for 1 hour at 4°C to reduce nonspecific binding.
C. Immunoprecipitation & Washes
- Materials: High-specificity, validated antibody (see Toolkit), magnetic beads, low-salt wash buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS), high-salt wash buffer (as above with 500 mM NaCl), LiCl wash buffer (10 mM Tris-HCl pH 8.0, 250 mM LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA), TE buffer.
- Method:
- Incubate pre-cleared chromatin with antibody (1-10 μg) overnight at 4°C. Antibody quality is the single largest factor affecting enrichment.
- Add 40 μL pre-blocked beads, incubate 2-4 hours.
- Wash beads sequentially for 5 min each: 2x low-salt, 1x high-salt, 1x LiCl, 2x TE buffer. Stringent washes reduce background.
D. Elution, Reverse Cross-linking & Purification
- Materials: Elution buffer (50 mM NaHCO₃, 1% SDS), Proteinase K, RNase A, NaCl, QIAquick PCR Purification Kit.
- Method:
- Elute chromatin from beads in 200 μL elution buffer, 65°C for 15 min with shaking. Combine with Input control.
- Reverse cross-link by adding 200 mM NaCl (final) and incubating overnight at 65°C.
- Add Proteinase K and RNase A, incubate 2 hours at 55°C.
- Purify DNA with spin columns, elute in 30-50 μL EB buffer.
E. Library Prep & Sequencing
- Materials: NEBNext Ultra II DNA Library Prep Kit, size selection beads (e.g., SPRIselect), appropriate sequencing primers.
- Method: Follow kit protocol. Use size selection to remove adapter dimers and large fragments. Sequence with ≥5 million non-duplicate reads for TF ChIP-seq on a platform like Illumina NovaSeq.
Diagnostic Pathways and Solutions
The relationship between root causes, symptoms, and corrective actions is depicted in the following diagnostic workflow.
Diagnostic Workflow for Poor ChIP-seq Signal
Computational Remediation Post-Sequencing
When experimental flaws are irreversible, computational methods can partially salvage data.
A. Adapter & Quality Trimming
- Tool: Trim Galore! or cutadapt.
- Command (Trim Galore):
trim_galore --paired --nextera -q 20 --length 25 -o ./output R1.fastq.gz R2.fastq.gz
B. Advanced Background Subtraction & Peak Calling
- Tool: MACS2 with a matched control (Input or IgG).
- Command (with stringent settings):
macs2 callpeak -t ChIP.bam -c Control.bam -f BAMPE -g hs -n Output --keep-dup all -q 0.01 --bw 300Note:--bwsets bandwidth to model sharper TF peaks.
C. Blacklist Region Filtering
- Resource: ENCODE Blacklist (hg38, mm10, etc.).
- Tool: bedtools intersect.
- Command:
bedtools intersect -v -a peaks.narrowPeak -b blacklist.bed > filtered_peaks.narrowPeak
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Robust TF ChIP-seq
| Reagent / Material | Function & Critical Role | Example Product / Note |
|---|---|---|
| High-Specificity Antibody | Binds target TF with minimal off-target interaction; the most critical reagent. | Validated ChIP-seq grade from Diagenode, Cell Signaling Technology, or Abcam. Always check for published datasets. |
| Protein A/G Magnetic Beads | Efficient capture of antibody-bound complexes, enabling stringent washing. | Dynabeads (Thermo Fisher), Sera-Mag beads. Superior to agarose beads for wash efficiency. |
| Cross-linking Reagent | Reversibly fixes protein-DNA interactions. | Ultrapure formaldehyde (Thermo Fisher, 28906). Methanol-free, fresh aliquots prevent over/under-fixation. |
| Chromatin Shearing Device | Fragments chromatin to optimal size (100-500 bp) for resolution. | Covaris S2/S220 (ultrasonication) or Bioruptor (diagenode). Consistent shearing is key. |
| Size Selection Beads | Purifies and size-selects libraries, removing primers/dimers. | SPRIselect (Beckman Coulter) or AMPure XP beads. Ratios are critical for fragment selection. |
| Library Prep Kit for Low Input | Converts low-yield ChIP DNA into sequencing libraries efficiently. | NEBNext Ultra II DNA Library Prep, SMARTer ThruPLEX. Optimized for <10 ng input. |
| qPCR Primers for Positive/Negative Genomic Loci | Pre- and post-ChIP quality control to assess enrichment fold-change. | Design primers for known binding site (positive) and gene desert (negative control). |
| RNase A & Proteinase K | Degrades RNA and proteins post-IP to purify DNA. | Molecular biology grade, RNase-free. Essential for clean DNA recovery. |
Diagnosing and remediating poor signal in TF ChIP-seq requires a systematic investigation of both the wet-lab protocol and computational pipeline. Low enrichment typically points to antibody or fixation issues, while high background implicates shearing or washing stringency. By adhering to rigorous QC protocols, utilizing validated reagents from the toolkit, and applying appropriate computational corrections, researchers can rescue studies and generate high-quality, publication-ready transcription factor binding data integral to the broader thesis of gene regulation analysis.
Within the comprehensive ChIP-seq data analysis workflow for transcription factor (TF) research, peak calling is the critical step that translates aligned sequence reads into genomic regions of putative protein-DNA interaction. A one-size-fits-all parameter set is insufficient due to the diverse biological behaviors of TFs. This technical guide details the rationale and methodology for parameter optimization tailored to TF-specific characteristics, ensuring accurate biological interpretation in research and drug discovery.
Transcription Factor Classification and Parameter Implications
Transcription factors exhibit distinct chromatin-binding behaviors, primarily categorized as Pioneer Factors, Classical Sequence-Specific TFs, and Co-factors/Chromatin Regulators. Their behavior dictates optimal peak-calling parameters.
Table 1: TF Behavioral Classification and Peak Characteristics
| TF Class | Binding Motif | Peak Shape | Genomic Distribution | Example TFs |
|---|---|---|---|---|
| Pioneer | Degenerate, broad | Broad, diffuse | Heterochromatic regions | FOXA1, PU.1 |
| Classical | Sharp, specific | Narrow, sharp | Promoters, Enhancers | p53, STAT1 |
| Co-factor | Variable (often indirect) | Mixed | Near other TF peaks | p300, MED1 |
Table 2: Key MACS2 Parameters for Different TF Behaviors
| Parameter | Function | Pioneer/ Broad TF Value | Classical/ Sharp TF Value | Rationale |
|---|---|---|---|---|
--bw (bandwidth) |
Smoothing window for model building | 300-500 bp | 100-200 bp | Matches the broader ChIP enrichment landscape. |
--mfold |
Range for model building | 5 100 | 10 30 | Broad regions have lower enrichment folds. |
--nomodel & --extsize |
Use fixed shift size | Often used (--extsize 200-300) | Rarely used | Overrides model for consistent broad peak detection. |
--qvalue (or -p) |
Significance threshold | 0.01 | 0.05 | Stricter threshold reduces false positives in noisy broad regions. |
--broad |
Enables broad peak calling | Yes | No | Critical for calling broad domains. |
--broad-cutoff |
Threshold for broad peaks | 0.1 | N/A | Relaxed cutoff for broad regions. |
Experimental Protocols for Parameter Calibration
Protocol: Empirical Optimization Using Spike-in Control
Purpose: To normalize for technical variation (e.g., antibody efficiency, total IP mass) and enable quantitative comparison of enrichment levels across experiments, which informs --mfold and -q settings.
Materials:
- ChIP sample from cells/tissue.
- Commercially available spike-in chromatin (e.g., from Drosophila melanogaster or S. cerevisiae) and corresponding species-specific antibody.
- Cross-linked chromatin from the spike-in organism.
- Paired-end sequencing platform.
Methodology:
- Spike-in Addition: Add a fixed, small amount (typically 1-10%) of exogenous spike-in chromatin to your experimental ChIP sample after sonication but before immunoprecipitation.
- Sequencing & Alignment: Sequence the library. Align reads simultaneously to the primary (e.g., human, hg38) and spike-in (e.g., dm6) reference genomes using an aligner like BWA-MEM or Bowtie2.
- Enrichment Calculation: Perform peak calling separately on the primary and spike-in alignments. Calculate the FRiP (Fraction of Reads in Peaks) for both.
- Normalization Factor: Derive a scaling factor based on the spike-in FRiP ratio between samples. This factor adjusts for global differences in ChIP efficiency.
- Parameter Adjustment: Apply the scaling factor to understand true biological signal strength. If the normalized enrichment is consistently low (e.g., <5-fold), use a more permissive
--mfold(e.g., 5 50) for model building.
Protocol: Motif Recovery Validation
Purpose: To assess peak-calling specificity by measuring the frequency of the known cognate motif within called peaks, optimizing the -q/-p cutoff.
Materials:
- List of called peaks (BED or narrowPeak format).
- Reference genome FASTA file.
- Known Position Weight Matrix (PWM) for the TF (from JASPAR, CIS-BP).
- Software: HOMER, MEME-ChIP, or FIMO.
Methodology:
- Peak Subsetting: Generate multiple peak lists from the same experiment using a range of significance cutoffs (e.g., q-value: 0.001, 0.01, 0.05, 0.1).
- Motif Scanning: For each peak list, extract genomic sequences (e.g., ±100 bp from summit) and scan for the known TF motif using
findMotifsGenome.pl(HOMER) orfimo(MEME Suite). - Calculate Recovery Rate: Determine the percentage of peaks containing a significant (p < 1e-4) motif hit for each cutoff.
- Plot & Determine Optimum: Plot q-value cutoff vs. motif recovery rate. The optimal cutoff is often at the "elbow" of the curve, balancing specificity and sensitivity.
Table 3: Example Motif Recovery Results
| Q-value Cutoff | Number of Peaks | Peaks with Motif (%) | Recommended Use Case |
|---|---|---|---|
| 0.001 | 1,250 | 85% | Ultra-high confidence, core set for strict validation. |
| 0.01 | 5,780 | 78% | Optimal balance for most sharp TF analyses. |
| 0.05 | 12,450 | 65% | Sensitive set for genome-wide or co-factor analysis. |
| 0.1 | 18,900 | 52% | Overly permissive; high false positive rate. |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents and Tools for Optimized ChIP-seq
| Item | Function | Example/Supplier |
|---|---|---|
| Spike-in Chromatin & Antibody | Normalizes for technical variation between samples. | Drosophila S2 chromatin & anti-H2Av (Active Motif, #61686). |
| Validated ChIP-Grade Antibody | Specific immunoprecipitation of the target TF. | Cell Signaling Technology, Abcam, Diagenode. |
| Magnetic Protein A/G Beads | Efficient capture of antibody-antigen complexes. | Dynabeads (Thermo Fisher). |
| High-Fidelity Library Prep Kit | Minimizes bias during NGS library construction. | KAPA HyperPrep (Roche) or NEBNext Ultra II (NEB). |
| qPCR Primers for Positive/Negative Genomic Loci | Validates ChIP enrichment prior to sequencing. | Design primers for known binding sites and inert regions. |
| Peak Caller Software | Identifies statistically significant enrichment regions. | MACS2 (broad/narrow), SPP, HOMER. |
| Motif Analysis Suite | Discovers de novo or matches known motifs in peaks. | HOMER, MEME-ChIP, RSAT. |
Visualization of Workflows and Relationships
Diagram 1: Peak Calling Optimization Workflow (98 chars)
Diagram 2: TF Behavior Dictates Peak Calling Parameters (91 chars)
Integrating behavioral classification of TFs with empirical calibration protocols is not an optional refinement but a core component of a rigorous ChIP-seq workflow. By systematically adjusting peak-calling parameters—guided by spike-in normalization and motif recovery validation—researchers can derive accurate, high-confidence binding profiles. This precision is fundamental for downstream analyses, such as identifying disease-associated regulatory networks or evaluating drug-mediated changes in TF activity, thereby directly impacting the efficacy and safety of therapeutic development.
Within a comprehensive ChIP-seq data analysis workflow for transcription factor research, addressing technical artifacts is a critical preprocessing step. Two pervasive sources of noise are PCR duplicates and reads aligning to blacklisted regions. Their proper identification and mitigation are fundamental to ensuring the biological fidelity of downstream analyses, such as peak calling and motif discovery, which underpin mechanistic studies in drug development.
PCR Duplicates in ChIP-seq
Definition and Origin
PCR duplicates are sequences originating from the same original DNA fragment due to clonal amplification during the library preparation's polymerase chain reaction (PCR) step. In ChIP-seq, they can artificially inflate the signal strength at specific genomic loci, leading to false-positive peak calls.
Quantitative Impact
The following table summarizes typical rates and impacts of PCR duplicates in standard transcription factor ChIP-seq experiments.
Table 1: Characteristics and Impact of PCR Duplicates in TF ChIP-seq
| Metric | Typical Range | Implication for Analysis |
|---|---|---|
| Duplicate Rate | 10-30% (varies by sequencing depth & protocol) | High rates (>50%) suggest low complexity libraries. |
| Signal Skew | Can account for >70% of reads at a peak summit | Leads to overestimation of binding affinity. |
| Peak Caller Sensitivity | False positives increase ~15-25% if not removed | Compromises specificity of binding site identification. |
Protocol for Identification and Removal
Method: MarkDuplicates (Picard Tools/GATK)
- Input: Coordinate-sorted BAM file (aligned reads).
- Process: The tool identifies duplicate reads defined as those with:
- Same 5' alignment start position (for paired-end, both ends).
- Same outer alignment coordinates.
- Same unique molecular identifier (UMI), if incorporated in the protocol.
- Algorithm: Retains a single primary alignment (highest base quality) and marks all others as duplicates in the BAM flag.
- Output: A new BAM file with duplicate flags set, plus a metrics file (summarized in Table 2).
- Downstream: Marked reads are typically excluded during peak calling.
Table 2: Example Output Metrics from Picard MarkDuplicates
| Library Metric | Value | Interpretation |
|---|---|---|
| UNPAIREDREADSEXAMINED | 1,450,200 | Total reads processed. |
| READPAIRSEXAMINED | 4,850,500 | Total read pairs processed. |
| PERCENT_DUPLICATION | 22.5% | Fraction of reads considered duplicates. |
| ESTIMATEDLIBRARYSIZE | 12,450,000 | Estimated unique DNA fragments. |
Title: ChIP-seq PCR Duplicate Removal Workflow
Blacklisted Regions in the Genome
Definition and Origin
Blacklisted regions are genomic areas with consistently high, unstructured signals across experimental types and cell lines. They arise from:
- Artifactual signal: from repetitive sequences, satellite DNA, or poor mappability.
- Structured artifacts: from ultra-high signal in regions like telomeres and centromeres. For transcription factor analysis, peaks in these regions are almost always non-biological.
Standardized Blacklists
Consortium-curated lists are essential. The most widely used is the ENCODE Blacklist for model organisms (hg19, hg38, mm9, mm10).
Table 3: ENCODE Blacklist Regions for Key Organisms
| Genome Build | Total Blacklisted Bases | Number of Regions | Primary Genomic Features |
|---|---|---|---|
| hg38 (Human) | ~162 Mb | 1640 | Centromeres, telomeres, satellite repeats |
| mm10 (Mouse) | ~151 Mb | 1641 | High-density repeat regions |
| dm6 (Fly) | ~16 Mb | 226 | Artifact-prone heterochromatin |
Protocol for Region Filtering
Method: BEDTools intersect
- Input:
- BED file of called peaks (e.g., from MACS2).
- BED file of blacklisted regions for the correct genome build.
- Command:
bedtools intersect -a peaks.bed -b blacklist.bed -v > peaks_filtered.bed- The
-vflag reports only entries in-athat do not overlap with-b.
- The
- Output: A filtered BED file with all peaks falling within blacklisted regions removed.
- Best Practice: Some workflows also filter the aligned BAM file before peak calling to prevent spurious alignments from influencing background models.
Title: Logical Decision Tree for Peak Blacklist Filtering
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Tools and Reagents for Artifact Mitigation
| Item Name | Provider/Example | Function in Addressing Artifacts |
|---|---|---|
| High-Fidelity PCR Enzyme | KAPA HiFi, Q5 Hot Start | Minimizes PCR bias and errors during library amplification, reducing duplicate-eligible fragments. |
| Unique Molecular Identifiers (UMIs) | NEBNext Unique Dual Index UMI Adapters | Tags each original DNA fragment with a random barcode, allowing true duplicates (same UMI) to be distinguished from PCR duplicates. |
| ENCODE Blacklist BED Files | ENCODE Consortium Portal | Provides standardized, curated lists of problematic genomic regions to filter out artifactual signals. |
| Picard Tools | Broad Institute | The industry-standard Java suite containing MarkDuplicates for duplicate identification and marking. |
| BEDTools | Quinlan Lab | A flexible Swiss-army-knife for genomic arithmetic; used to filter peaks/BAM files against blacklists. |
| MACS2 Peak Caller | Zhang Lab | Incorporates a --keep-dup parameter to control how duplicates are used during statistical modeling of peaks. |
| SAMtools | Li Lab | Used for manipulating BAM files (sorting, indexing) which is a prerequisite for duplicate marking and filtering. |
Integrated Workflow in a ChIP-seq Analysis Pipeline
The handling of these artifacts is sequential and integrated into the early stages of data processing.
Title: Artifact Mitigation in ChIP-seq Preprocessing
In the context of a robust ChIP-seq workflow for transcription factor research, systematic removal of PCR duplicates and filtering of blacklisted regions are non-negotiable steps for data integrity. These procedures directly enhance the signal-to-noise ratio, leading to a more accurate and reliable set of binding sites. This accuracy is paramount for subsequent functional validation and the identification of targetable pathways in drug discovery, ensuring that resources are focused on biologically relevant mechanisms.
The analysis of transcription factor (TF) binding sites using Chromatin Immunoprecipitation followed by sequencing (ChIP-seq) is a cornerstone of functional genomics. A persistent challenge in this workflow is the accurate capture and identification of binding events for TFs that exhibit weak affinity or transient, dynamic interactions with DNA. These interactions are often biologically significant but are prone to being lost as noise due to low signal-to-noise ratios, non-specific antibody binding, and limitations in crosslinking efficiency. This technical guide outlines strategies integrated at both wet-lab and computational stages of the ChIP-seq pipeline to enhance the specificity for such elusive TF-DNA interactions.
Core Challenges & Quantitative Data
The difficulties in studying weak/transient TFs are quantifiable. The following table summarizes key parameters compared to stable TF interactions.
Table 1: Quantitative Comparison of TF Interaction Types in ChIP-seq
| Parameter | Stable/High-Affinity TF Interactions | Weak/Transient TF Interactions |
|---|---|---|
| Typical Residence Time | > 30 seconds | < 10 seconds |
| Crosslinking Efficiency | High (5-10%) | Very Low (<1-2%) |
| Peak Sharpness (Avg. Width) | Narrow (< 200 bp) | Very Broad (> 1000 bp) or undetectable |
| Signal-to-Noise Ratio (SNR) | High (> 10:1) | Low (< 3:1) |
| Optimal Sequencing Depth | 20-40 million reads | 50-100+ million reads |
| % of Reads in Peaks (FRiP) | 5-20% | Often < 1-2% |
Experimental Protocol Enhancements
Optimized Crosslinking & Chromatin Preparation
Protocol: Double Crosslinking for Transient TFs
- Reagents: Disuccinimidyl glutarate (DSG), Formaldehyde (37%), Glycine (2.5 M), Lysis buffers.
- Procedure:
- Wash cells with cold PBS.
- Add DSG (2 mM final concentration in PBS) and incubate for 45 min at room temperature to stabilize protein-protein interactions.
- Quench DSG with 100 mM Tris-HCl (pH 7.5) for 5 min.
- Add Formaldehyde (1% final concentration) and incubate 10 min at room temperature to fix protein-DNA interactions.
- Quench with 125 mM Glycine for 5 min.
- Proceed with standard cell lysis and chromatin shearing via sonication. Aim for fragment sizes of 150-300 bp.
Advanced Immunoprecipitation Strategies
Protocol: Carrier-Assisted ChIP (caChIP)
- Principle: Uses a high-abundance, epitope-tagged "carrier" TF expressed at low levels to improve precipitation kinetics and recovery of low-abundance complexes.
- Procedure:
- Generate cell line stably expressing a FLAG-tagged version of the target TF at near-endogenous levels (or use a known high-affinity TF as carrier).
- Perform crosslinking and chromatin preparation as above.
- During IP, use an anti-FLAG antibody alongside the target TF antibody. The FLAG-TF acts as a carrier during bead capture.
- Wash stringently (e.g., high-salt wash, LiCl wash) to reduce non-specific background.
- Elute and reverse crosslinks. Analyze DNA specifically bound by the endogenous TF via qPCR or sequencing.
Nuclease-Based Alternatives: CUT&RUN and CUT&Tag
Protocol: CUT&Tag for Native Conditions
- Principle: Uses a protein A-Tn5 fusion protein targeted by an antibody to cleave and tag genomic regions bound by the TF in situ, without crosslinking.
- Procedure:
- Permeabilize intact nuclei with digitonin.
- Incubate with primary antibody against the target TF (use at 2-5x standard ChIP concentration).
- Incubate with secondary antibody (if primary is not from guinea pig or rabbit).
- Bind pA-Tn5 fusion protein to the antibody complex.
- Activate Tn5 with Mg2+ to perform targeted tagmentation at the binding site.
- Extract and purify DNA, then amplify with indexed primers for sequencing. This method is highly sensitive for low-abundance factors.
Computational & Analytical Refinements
Post-sequencing, specialized bioinformatics tools are crucial.
Table 2: Computational Tools for Weak TF Signal Analysis
| Tool Name | Primary Function | Key Parameter for Weak TFs |
|---|---|---|
| MACS3 (broad peak calling) | Peak calling | Use --broad flag, lower -q value cutoff (e.g., 0.1). |
| SEACR | Peak calling from sparse data | Uses control to define threshold via AUC; effective for low SNR. |
| S3V2 | Identifies variable-length peaks | Models shape variation, good for diffuse signals. |
| ChIP-Rx | Normalization with spike-in chromatin | Uses exogenous D. melanogaster chromatin to normalize for technical variation. |
| NF-CORE ChIP-seq | Standardized pipeline | Incorporates multiple callers and quality metrics for robust analysis. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Studying Weak/Transient TF Interactions
| Reagent/Material | Function & Rationale |
|---|---|
| Disuccinimidyl Glutarate (DSG) | A reversible amine-reactive crosslinker for protein-protein interactions, stabilizing transient complexes prior to formaldehyde fixation. |
| FLAG Epitope Tag System | Allows for high-affinity immunoprecipitation of low-abundance TFs when expressed as a fusion carrier or target. |
| pA-Tn5 Fusion Protein | Essential enzyme for CUT&RUN/CUT&Tag, enabling antibody-directed integration of sequencing adapters at binding sites with low background. |
| Digitonin | A mild detergent for nuclear permeabilization in CUT&RUN/Tag, preserving native chromatin state. |
| D. melanogaster Chromatin (Spike-in) | Exogenous chromatin added prior to IP for quantitative normalization between samples, correcting for IP efficiency differences. |
| High-Specificity Antibodies (Monoclonal/ Recombinant) | Minimizes non-specific background; recombinant antibodies offer superior lot-to-lot consistency for low-signal applications. |
| Methylcellulose | Used in some protocols to stabilize nuclei and reduce diffusion during in situ assays like CUT&Tag. |
Visualizing Strategies and Workflows
Strategy Selection for Weak TF ChIP
Co-factor Role in Stabilizing TF Binding
Batch Effect Correction and Normalization Across Multiple Samples
In ChIP-seq analysis for transcription factor (TF) binding studies, batch effects are systematic non-biological variations introduced during sample handling, sequencing runs, reagent lots, or personnel changes. These artifacts can confound true biological signals, leading to false positives or negatives when comparing samples across experiments. Effective batch effect correction is therefore a critical step in any robust ChIP-seq workflow, ensuring that observed differences in peak calls and binding intensities accurately reflect underlying biology rather than technical noise.
Batch effects arise from multiple stages of the ChIP-seq protocol.
Table 1: Common Sources of Batch Effects in TF ChIP-seq Workflows
| Protocol Stage | Specific Source | Potential Impact on Data |
|---|---|---|
| Cell Culture & Crosslinking | Passage number, confluency, crosslinking time/temp | Variation in chromatin accessibility & fixation efficiency |
| Immunoprecipitation | Antibody lot, incubation time, washing stringency | Differences in enrichment specificity and yield |
| Library Preparation | Kit version, PCR cycle number, personnel | Biases in fragment size selection & amplification |
| Sequencing | Flow cell, lane, cluster density, chemistry version | Differences in read depth, quality scores, and GC bias |
Pre-Normalization: Quality Assessment and Read Alignment
Before correction, data quality must be assessed.
Experimental Protocol 1: Cross-Correlation Analysis for TF ChIP-seq
- Purpose: Assess signal-to-noise and fragment size.
- Steps:
- Align reads using a spliced aligner (e.g., Bowtie2, BWA) with appropriate parameters for short reads.
- Filter aligned reads for mapping quality (MAPQ > 10) and remove duplicates using tools like Picard.
- Use the
phantompeakqualtoolspackage to calculate the cross-correlation profile. - Extract metrics: Normalized Strand Cross-Correlation coefficient (NSC) and Relative Strand Cross-Correlation coefficient (RSC). NSC > 1.05 and RSC > 0.8 indicate good quality for TF experiments.
- Key Reagents: Nuclease-free water, Tris-EDTA buffer.
Normalization Methods to Account for Technical Variation
Normalization aims to make samples comparable by adjusting for technical biases like sequencing depth.
Table 2: Common Normalization Methods for ChIP-seq Data
| Method | Principle | Use Case | Tool/Implementation |
|---|---|---|---|
| Reads Per Million (RPM/CPM) | Scales reads by total mapped reads per sample. | Initial assessment, depth adjustment. | BedTools, deepTools bamCoverage |
| Trimmed Mean of M-values (TMM) | Uses a reference sample to calculate scaling factors based on most stable peaks. | Between-sample normalization when global differences are small. | edgeR R package |
| Median Ratio Normalization | Assumes most peaks are not differentially bound. Calculates a size factor as the median of ratios to a geometric mean. | Suitable for experiments with many shared peaks. | DESeq2 R package |
| Peak-Based Quantile | Equalizes the distribution of signal intensities across called peak regions. | For focused analysis on pre-defined peak sets. | limma, ChIPseqSpikeInFree |
Batch Effect Correction Algorithms
These methods model and remove batch-specific variation after normalization.
Experimental Protocol 2: Batch Correction using ComBat-seq (for Count Data)
- Purpose: Remove batch effect while preserving biological signal using an empirical Bayes framework.
- Steps:
- Generate a consensus peak set across all samples (e.g., using
MACS2callpeak on pooled reads). - Count reads in each consensus peak for every sample to create a count matrix.
- Define a batch variable (e.g., sequencing date) and optional biological covariates (e.g., condition).
- Run ComBat-seq from the
svaR package:adjusted_counts <- ComBat_seq(count_matrix, batch=batch_var, group=condition). - Use adjusted counts for downstream differential binding analysis.
- Generate a consensus peak set across all samples (e.g., using
Experimental Protocol 3: Batch Correction using RUV (Remove Unwanted Variation)
- Purpose: Use control regions (e.g., non-differential peaks, spike-ins) to estimate and remove unwanted factors.
- Steps:
- Identify a set of "negative control" peaks expected not to show differential binding across key biological conditions.
- Perform an initial normalization (e.g., RPM).
- Apply
RUVgorRUVsfrom theRUVSeqR package, specifying the control peaks and the number of unwanted factors (k) to remove. - The residuals from the RUV model are the batch-corrected signals.
Validation of Correction Efficacy
Correction success must be validated.
- Principal Component Analysis (PCA): Visualize sample clustering before/after correction. Biological replicates should cluster tightly, and separation should be driven by condition, not batch.
- Distribution Inspection: Overlaid density plots of signal intensity should show aligned distributions across batches post-correction.
Title: ChIP-seq Batch Effect Correction and Validation Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Controlled ChIP-seq Studies
| Item | Function | Example/Note |
|---|---|---|
| Spike-in Chromatin | External control for normalization across batches. | Drosophila chromatin (e.g., SNAP-Chip) or synthetic nucleosomes. |
| Commercial Control Antibodies | Positive (e.g., H3K4me3) and negative (IgG) controls for IP efficiency. | Essential for assessing protocol performance per batch. |
| Crosslinking Reagents | Formaldehyde (1%) for DNA-protein fixation. | Consistency in lot and quenching (glycine) is critical. |
| Magnetic Protein A/G Beads | Uniform capture of antibody-bound complexes. | Bead lot consistency minimizes variability. |
| Certified Low-DNA Enzyme Kits | For end repair, A-tailing, and adapter ligation. | Kit lot matching reduces library prep bias. |
| Indexed Adapter Kits | Multiplexing samples within a sequencing lane. | Balanced index use across batches minimizes lane effects. |
| Phusion HF Polymerase | High-fidelity amplification of library fragments. | Consistent PCR cycle number is vital. |
| Bioanalyzer/Tapestation Kits | Quality control of library fragment size distribution. | Used pre-sequencing to ensure batch similarity. |
Integrated Workflow and Best Practices
A recommended integrated workflow is:
- Design: Randomize samples across batches where possible.
- QC: Perform cross-correlation and FRiP analysis per sample.
- Alignment & Peak Calling: Use consistent parameters; generate a consensus peak set.
- Normalization: Apply depth normalization (e.g., via
edgeRorDESeq2). - Correction: Apply a batch correction method (e.g.,
ComBat-seq) using known batch variables. - Validation: Inspect PCA plots and correlation matrices post-correction.
Conclusion: In ChIP-seq studies for transcription factor biology, rigorous batch effect correction is not optional but a fundamental component of reproducible research. By systematically implementing the normalization and correction strategies outlined, researchers can ensure that conclusions about TF binding dynamics are biologically accurate and technically sound.
Within the context of a comprehensive ChIP-seq data analysis workflow for transcription factor (TF) research, the selection of appropriate statistical thresholds for peak calling is a pivotal step. This decision directly influences the downstream biological interpretation, affecting the identification of bona fide TF binding sites. This guide provides an in-depth technical examination of how to balance sensitivity (true positive rate) and the False Discovery Rate (FDR) to optimize discovery in TF ChIP-seq experiments.
Core Statistical Concepts in ChIP-seq Analysis
Sensitivity (Recall, True Positive Rate)
Sensitivity measures the proportion of actual binding sites correctly identified by the peak caller. In ChIP-seq, a high sensitivity minimizes false negatives, ensuring a more complete catalog of TF binding events, which is crucial for understanding regulatory networks.
False Discovery Rate (FDR)
FDR is the expected proportion of false positives among all peaks called. Controlling the FDR (e.g., at 1% or 5%) is essential for the reliability of downstream analyses, such as motif discovery and pathway enrichment.
The Precision-Recall Trade-off
Increasing sensitivity typically requires accepting a higher FDR, and vice versa. The optimal balance is experiment-specific and depends on the biological question, TF abundance, and data quality.
Quantitative Comparison of Thresholding Approaches
The following table summarizes common statistical measures and their impact on sensitivity and FDR.
Table 1: Statistical Measures and Their Implications in ChIP-seq Peak Calling
| Measure/Threshold | Typical Range | Impact on Sensitivity | Impact on FDR | Primary Use Case |
|---|---|---|---|---|
| q-value (FDR-adjusted p) | < 0.01 - 0.05 | Lower threshold increases sensitivity | Directly controlled; lower q-value lowers FDR | Standard for final high-confidence peak lists |
| p-value | < 1e-5 | Lower threshold increases stringency, lowers sensitivity | Indirect control; lower p-value typically lowers FDR | Initial filtering; less reliable than q-value |
| Fold Enrichment (over control) | > 5 - 10 | Higher threshold decreases sensitivity | Higher threshold generally decreases FDR | Filtering broad or diffuse peaks; requires good control |
| Peak Score (e.g., -log10(p)) | Varies by caller | Higher score decreases sensitivity | Higher score decreases FDR | Caller-specific ranking metric |
Detailed Experimental Protocol: Validating Threshold Choices
A systematic approach to threshold selection involves benchmarking against known binding sites or orthogonal validation.
Protocol: Empirical Optimization of q-value Threshold Using a Validation Dataset
Objective: To determine the optimal q-value cutoff that maximizes the confirmation rate of ChIP-seq peaks in an independent validation assay (e.g., EMSA or TF perturbation RNA-seq).
Materials:
- ChIP-seq peak calls at varying q-value thresholds (e.g., 0.001, 0.01, 0.05, 0.1).
- A set of known positive binding regions (e.g., from public databases like ChIP-Atlas or validated sites from literature).
- A set of known negative regions (e.g., silent genomic regions, or peaks from an irrelevant IgG control).
Procedure:
- Generate Thresholded Peak Sets: Using your peak caller (e.g., MACS2), output peak lists filtered at different q-value (FDR) cutoffs.
- Overlap with Reference Sets: For each thresholded list, calculate the overlap with the known positive and known negative sets using tools like
bedtools intersect. - Calculate Performance Metrics: For each threshold, compute:
- True Positives (TP): Peaks overlapping known positive regions.
- False Positives (FP): Peaks overlapping known negative regions or non-overlapping with positives.
- Sensitivity/Recall: TP / (Total known positives).
- Precision: TP / (TP + FP).
- Plot & Determine Optimum: Construct a Precision-Recall curve by plotting precision against recall for each threshold. The optimal threshold is often at the "elbow" of the curve or based on the project's need for high precision (low FDR) or high recall.
- Cross-validate: Perform the same analysis on biological replicates to ensure robustness.
Visualizing the Analysis Workflow and Decision Logic
ChIP-seq Analysis and Threshold Decision Workflow
Decision Logic for Selecting Statistical Thresholds
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Research Reagent Solutions for ChIP-seq Threshold Validation
| Reagent / Material | Function in Threshold Validation | Key Consideration |
|---|---|---|
| Validated Antibody (for TF of interest) | High-specificity antibody is critical for generating the primary ChIP-seq data to be thresholded. | Validation by knockout/knockdown is ideal to assess off-target peaks. |
| IgG Isotype Control | Provides a nonspecific antibody control to assess background noise. Essential for defining FDR. | Must match the host species and immunoglobulin class of the primary antibody. |
| PCR Purification Kit | For purifying ChIP-enriched DNA before library preparation. Clean DNA improves library complexity. | Minimize size selection bias; elute in low-EDTA TE buffer or nuclease-free water. |
| High-Sensitivity DNA Assay Kit (e.g., Qubit) | Accurate quantification of low-concentration ChIP DNA is essential for successful library prep. | Fluorometric assays are superior to absorbance (Nanodrop) for low-concentration samples. |
| Library Preparation Kit (with dual-size selection) | Converts ChIP DNA to sequencing-ready libraries. Dual-size selection improves peak resolution. | Choose kits optimized for low-input DNA. Include UMIs to mitigate PCR duplicates. |
| Synthetic Spike-in DNA (e.g., from Drosophila) | Added to ChIP reactions before sequencing to normalize samples and compare sensitivity across experiments. | Use a non-homologous genome (e.g., D. melanogaster for human samples) and a corresponding antibody. |
| EMSA/Gel Shift Kit | For orthogonal validation of specific TF-DNA interactions from called peaks. Confirms precision (low FDR). | Useful for testing a subset of high-scoring and medium-scoring peaks. |
| qPCR Reagents & Primers | For qPCR validation of enrichment at specific loci versus negative control regions. Assesses sensitivity. | Design primers for top peaks, random peaks, and negative genomic regions. |
Computational Resource Management for Large-scale ChIP-seq Projects
Within the broader thesis on ChIP-seq data analysis workflows for transcription factor research, the efficient management of computational resources emerges as a critical bottleneck. As projects scale to encompass hundreds to thousands of samples—common in drug discovery and comparative studies—the demands on storage, processing power, and workflow orchestration increase exponentially. This technical guide details the core considerations and methodologies for managing these resources effectively, ensuring reproducible, timely, and cost-effective research.
Computational Resource Landscape for Large-scale ChIP-seq
Large-scale ChIP-seq projects involve sequential and parallel processing stages, each with distinct resource profiles. The primary phases are: 1) Raw Data Acquisition & Storage, 2) Primary Analysis (Alignment), 3) Secondary Analysis (Peak Calling & QC), and 4) Tertiary Analysis (Comparative & Integrative Analysis).
Table 1: Estimated Computational Load per Sample (Human Genome, ~50M reads)
| Analysis Phase | CPU Cores (Recommended) | Wall-clock Time (hrs) | Peak RAM (GB) | Storage I/O | Output Size |
|---|---|---|---|---|---|
| FASTQ QC | 4-8 | 0.5-1 | 4-8 | High Read | ~1 GB |
| Alignment | 8-16 | 2-4 | 12-16 | Very High | ~15-20 GB |
| Post-Alignment QC | 4 | 0.5 | 8 | Medium | ~0.5 GB |
| Peak Calling | 4-8 | 1-3 | 8-12 | Medium | ~0.1-0.5 GB |
| Downstream Analysis | 4-32* | 1-48* | 16-64* | Variable | Variable |
*Highly dependent on the specific tool and comparison complexity.
Table 2: Aggregate Storage Requirements for Project Scale
| Project Scale | Samples | Raw Data (FASTQ) | Processed Data (BAM, etc.) | Total Estimated (w/ redundancy) |
|---|---|---|---|---|
| Medium | 50 | 2-3 TB | 1-2 TB | 5-6 TB |
| Large | 500 | 20-30 TB | 10-15 TB | 50-70 TB |
| Very Large | 5000 | 200-300 TB | 100-150 TB | 0.5-1 PB |
Core Methodologies for Efficient Resource Management
Workflow Orchestration with HPC & Cloud
Effective management requires a robust workflow manager to handle job scheduling, dependency resolution, and failure recovery.
Protocol: Implementing a Nextflow Pipeline for Scalable ChIP-seq Analysis
- Define Process Modules: Create separate processes for each tool (e.g.,
fastqc,trim_galore,bwa_mem,macs2). Specify required CPU, memory, and time limits within each process definition. - Channel Design: Use input channels to supply sample sheets and reference genomes. Emit output channels for BAM files, peak files, and QC reports.
- Configuration Profiles: Establish separate configuration profiles (
conf/hpc.config,conf/cloud.config) to abstract execution environment details. Specify executor (Slurm, AWS Batch), queue parameters, and container technology (Docker/Singularity). - Checkpointing & Resumption: Leverage Nextflow's built-in resume capability (
-resume) to continue from the last successfully executed process after a failure or pause. - Resource Monitoring: Integrate with reporting tools like
Traceor custom scripts to log CPU, memory, and storage usage per process for optimization.
Data Lifecycle Management Protocol
A tiered storage strategy is essential for cost containment.
Protocol: Implementing a Tiered Storage Strategy
- Hot Storage (NVMe/SSD): Reserve for active processing of the current batch of samples (e.g., 10-20% of total project data). Configure workflow to stage input data here and write temporary files.
- Warm Storage (High-performance NAS): House all processed data (BAM, peaks) for active analysis and visualization. Data remains here for the project's duration.
- Cold Storage (Object Storage/ Tape): Archive raw FASTQ files and final project results after completion. Use lifecycle policies to automatically transition data from warm to cold storage after 6-12 months of inactivity.
- Metadata Catalog: Maintain a database (e.g., SQLite, PostgreSQL) linking sample IDs, file paths across storage tiers, processing versions, and QC metrics.
Containerization for Reproducibility
Containerization packages software, libraries, and environment variables.
Protocol: Creating and Deploying Analysis Containers
- Dockerfile Creation: Start from a minimal base image (e.g.,
ubuntu:22.04). Use multi-stage builds to keep image size small. Install all dependencies (e.g.,samtools,deeptools,MACS2) via package managers (apt, conda, pip). - Version Tagging: Tag images with the workflow version (e.g.,
chipseq-pipeline:v1.2). Push to a container registry (Docker Hub, Amazon ECR, Google Container Registry). - Integration with Workflow: In the Nextflow configuration, enable Docker/Singularity and specify the container image for each process or globally.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Reagents for Large-scale ChIP-seq
| Item | Function & Purpose |
|---|---|
| Workflow Manager (Nextflow/Snakemake) | Orchestrates complex, multi-step analyses across diverse computing environments, ensuring reproducibility and scalability. |
| Container Technology (Docker/Singularity) | Encapsulates the complete software environment, eliminating "works on my machine" issues and enabling portability between HPC and cloud. |
| Cluster/Cloud Scheduler (Slurm/AWS Batch) | Manages job queues, allocates CPU/memory resources, and schedules jobs across distributed compute nodes. |
| Reference Genome Indexes (BWA/HISAT2) | Pre-built alignment indexes are critical for efficient read mapping; must be stored on high-I/O storage. |
| Pipeline Configuration Files | YAML/Config files that define resource requests, tool parameters, and execution paths for different project scales. |
| Metadata Management Database | Tracks samples, file locations, processing status, and QC outcomes, essential for project navigation and provenance. |
| QC Aggregation Tool (MultiQC) | Automatically compiles QC reports from multiple tools (FastQC, SAMtools, etc.) into a single HTML report for holistic assessment. |
Strategic Visualizations
Diagram 1: Computational resource management architecture for large-scale ChIP-seq.
Diagram 2: Core ChIP-seq workflow with resource profile per step.
Validating ChIP-seq Results and Comparative Analysis Across Conditions
Within the workflow of transcription factor (TF) research initiated by ChIP-seq analysis, candidate TF binding events and regulated genes require rigorous functional validation. This guide details three core in vitro and in vivo techniques—quantitative PCR (qPCR), Electrophoretic Mobility Shift Assay (EMSA), and Reporter Assays—essential for confirming and characterizing protein-DNA interactions and their transcriptional consequences.
Quantitative PCR (qPCR) for ChIP-seq Target Validation
Following ChIP-seq peak calling, qPCR is the primary method for validating enrichment at specific genomic loci. It provides quantitative, high-sensitivity confirmation of TF binding.
Detailed Protocol: ChIP-qPCR Validation
- Sample Preparation: Use DNA purified from your ChIP experiment (ChIP-DNA) and from a control immunoprecipitation (Input DNA).
- Primer Design: Design 18-22 bp primers with ~50-60% GC content, yielding amplicons of 80-150 bp centered on the peak summit. Include positive control (known binding site) and negative control (non-enriched genomic region) primers.
- Reaction Setup: Prepare a SYBR Green master mix containing DNA polymerase, dNTPs, and buffer. Aliquot into a qPCR plate. Add ChIP-DNA or diluted Input DNA to respective wells in triplicate. A standard 20 µL reaction contains 10 µL master mix, 2 µL primer pair mix (final concentration 500 nM each), 3 µL nuclease-free water, and 5 µL template DNA.
- qPCR Run: Use a two-step cycling protocol:
- Hold Stage: 95°C for 2 min (polymerase activation).
- 40 Cycles: 95°C for 15 sec (denaturation), 60°C for 1 min (annealing/extension).
- Include a melt curve stage (65°C to 95°C, increment 0.5°C) to verify amplicon specificity.
- Data Analysis: Calculate the percent input for each target locus: % Input = 2^(Ct[Input] - Ct[ChIP]) x Fd x 100%, where Fd is the Input dilution factor. Compare enrichment at target loci versus negative control regions.
Table 1: Typical ChIP-qPCR Results for Hypothetical Transcription Factor "X"
| Genomic Locus | ChIP Ct (Mean ± SD) | Input Ct (Mean ± SD) | % Input Enrichment | Validation Outcome |
|---|---|---|---|---|
| Positive Control (Known Site) | 24.5 ± 0.2 | 22.1 ± 0.1 | 5.9% | Confirmed |
| Candidate Peak 1 | 26.8 ± 0.3 | 23.0 ± 0.2 | 1.5% | Confirmed |
| Candidate Peak 2 | 31.2 ± 0.5 | 22.8 ± 0.1 | 0.2% | Not Confirmed |
| Negative Control Region | 32.1 ± 0.6 | 22.5 ± 0.1 | 0.1% | - |
Workflow for ChIP-seq Target Validation via qPCR
Electrophoretic Mobility Shift Assay (EMSA) forIn VitroBinding
EMSA, or gel shift assay, directly visualizes the physical interaction between a purified TF (or nuclear extract) and a labeled DNA probe containing the putative binding motif from ChIP-seq peaks.
Detailed Protocol: EMSA
- Probe Preparation: Anneal complementary single-stranded oligonucleotides spanning the binding motif to create a double-stranded probe. Label the 5' end with biotin or a fluorophore. Purify via gel electrophoresis or column.
- Protein Preparation: Use purified recombinant TF protein or prepared nuclear extract. Determine optimal protein concentration empirically (e.g., 0-500 ng per reaction).
- Binding Reaction: Incubate protein with 10-20 fmol of labeled probe in binding buffer (10 mM HEPES, 50 mM KCl, 1 mM DTT, 2.5% glycerol, 0.05% NP-40, 100 µg/mL BSA, 50 ng/µL poly(dI·dC)) for 20-30 minutes at room temperature. Include controls: probe alone, and competition with 100-200x molar excess of unlabeled wild-type or mutated probe.
- Electrophoresis: Load reactions onto a pre-run, non-denaturing 5-6% polyacrylamide gel in 0.5x TBE buffer at 100V for 60-90 minutes at 4°C.
- Detection: Transfer DNA to a positively charged nylon membrane via wet or semi-dry transfer. Crosslink (if using UV). Detect labeled probe using streptavidin-HRP/chemiluminescence (biotin) or direct fluorescence imaging.
Table 2: Key Research Reagents for EMSA
| Reagent / Solution | Function & Specification |
|---|---|
| Biotin-end-labeled DNA Probe | High-affinity binding site sequence from ChIP-seq peak; labeled for sensitive detection. |
| Recombinant TF Protein | Purified, active transcription factor; essential for specific shift confirmation. |
| Poly(dI·dC) | Non-specific competitor DNA; reduces background protein-nucleic acid interactions. |
| Non-denaturing PAGE Gel | 5-6% acrylamide:bis (29:1) in 0.5x TBE; matrix for separating protein-DNA complexes. |
| Nylon Membrane (+) Charge | For efficient transfer and immobilization of nucleic acids post-electrophoresis. |
| Chemiluminescent Substrate | (e.g., Luminol/Peroxide) Generates light signal for HRP-based detection of biotin probe. |
EMSA Principle: Protein Binding Retards Probe Migration
Reporter Assays for Functional Validation of Transcriptional Activity
Reporter assays determine if the TF binding event identified by ChIP-seq and validated by EMSA has a functional consequence on gene expression.
Detailed Protocol: Dual-Luciferase Reporter Assay
- Reporter Construct Cloning: Clone the genomic region containing the ChIP-seq peak (wild-type or mutated) upstream of a minimal promoter driving the firefly luciferase (luc) gene in a plasmid vector.
- Cell Seeding & Transfection: Seed mammalian cells (e.g., HEK293) in 24-well plates. Co-transfect each well with:
- Experimental Reporter: 400 ng of your firefly luciferase construct.
- Control Reporter: 40 ng of a Renilla luciferase plasmid (e.g., pRL-TK) for normalization.
- Effector Plasmid: 100 ng of a plasmid expressing your TF (or empty vector control). Use a transfection reagent (e.g., lipofectamine) according to manufacturer protocol.
- Incubation: Incubate cells for 24-48 hours to allow gene expression.
- Luciferase Measurement: Lyse cells with Passive Lysis Buffer. Use a Dual-Luciferase Reporter Assay System. In a luminometer tube, mix lysate with Luciferase Assay Reagent II (measures firefly luciferase), read luminescence. Then add Stop & Glo Reagent (quenches firefly, activates Renilla), read luminescence again.
- Data Analysis: Calculate the normalized relative light units (RLU): Firefly RLU / Renilla RLU. Plot the fold change relative to the empty vector control for each reporter construct.
Table 3: Sample Reporter Assay Data for TF "X" on Candidate Enhancers
| Reporter Construct (Insert) | Normalized RLU (Firefly/Renilla) Mean ± SEM | Fold Activation vs Control | Functional Outcome |
|---|---|---|---|
| Empty Vector (No Insert) | 1.0 ± 0.1 | 1.0 | Baseline |
| Positive Control (Strong Enhancer) | 15.3 ± 1.2 | 15.3 | Positive Control |
| Candidate Peak 1 Sequence (WT) | 8.7 ± 0.6 | 8.7 | Functional Enhancer |
| Candidate Peak 1 Sequence (Mut) | 1.2 ± 0.2 | 1.2 | Loss-of-Function |
| Candidate Peak 2 Sequence (WT) | 1.5 ± 0.3 | 1.5 | No Activity |
Dual-Luciferase Reporter Assay for Transcriptional Activity
Integrated Validation Workflow
These methods form a complementary, sequential validation pipeline stemming from initial ChIP-seq discovery.
Sequential Experimental Validation Pipeline
Within a comprehensive ChIP-seq data analysis workflow for transcription factor research, cross-platform validation is a critical step for ensuring the biological veracity of identified regulatory elements. While ChIP-seq identifies protein-DNA interaction sites, it benefits from orthogonal validation using open chromatin assays. ATAC-seq (Assay for Transposase-Accessible Chromatin) and DNase-seq (DNase I hypersensitive sites sequencing) are two predominant techniques for mapping chromatin accessibility. This guide details the methodology and rationale for integrating these datasets to validate and refine ChIP-seq-derived transcription factor binding sites and cis-regulatory elements, thereby strengthening downstream conclusions in drug discovery and mechanistic studies.
Core Principles of ATAC-seq and DNase-seq
ATAC-seq utilizes a hyperactive Tn5 transposase to simultaneously fragment and tag accessible genomic regions with sequencing adapters. DNase-seq employs the DNase I enzyme to cleave accessible DNA, followed by fragment end-capture and sequencing. Both map open chromatin, but with technical and practical differences.
Table 1: Quantitative Comparison of ATAC-seq and DNase-seq
| Feature | ATAC-seq | DNase-seq |
|---|---|---|
| Input Material | 500 - 50,000 nuclei/cells | 1 - 50 million nuclei/cells |
| Assay Time | ~3 hours hands-on, <1 day total | ~2 days hands-on, 3-4 days total |
| Primary Enzyme | Tn5 Transposase | DNase I Endonuclease |
| Fragment Size Profile | Periodic ~200-bp pattern (nucleosome positioning) | Continuous smear of fragment sizes |
| Sequence Bias | Moderate Tn5 sequence preference | Minimal sequence preference |
| Sensitivity | High (low cell input) | Moderate to High (requires more input) |
| Signal Resolution | Single-nucleotide (cut sites) | ~20-50 bp (cut clusters) |
| Multimodal Data | Nucleosome positioning inferred | Primarily accessibility only |
Integration Methodology for ChIP-seq Validation
Preprocessing and Peak Calling
Standardized preprocessing is essential for fair comparison.
- ATAC-seq Pipeline: Raw FASTQ → Adapter trim (e.g., Trimmomatic) → Alignment (BWA-MEM2/Bowtie2) → Remove mitochondrial reads and PCR duplicates → Shift + strand reads +4 bp, - strand reads -5 bp. Call peaks using MACS2.
- DNase-seq Pipeline: Raw FASTQ → Adapter trim → Alignment → Duplicate removal → Generate DNase I cleavage profiles (cut counts). Call hypersensitive sites (peaks) using F-Seq or MACS2.
Table 2: Recommended Peak-Calling Parameters
| Parameter | ATAC-seq (MACS2) | DNase-seq (F-Seq) |
|---|---|---|
| Bandwidth (-b) | 200 | 20 |
| p-value cutoff | 1e-5 | 1e-5 |
| Shift Size | Model-based | Not Applicable |
| Extension Size | Not Applicable | 600 |
Cross-Platform Peak Concordance Analysis
ChIP-seq peaks for a transcription factor (TF) should be enriched in accessible chromatin regions.
- Jaccard Index Calculation: Measures overlap between ATAC-seq and DNase-seq peak sets from the same sample.
J = (A ∩ D) / (A ∪ D), where A=ATAC peaks, D=DNase peaks. Values >0.2 indicate good technical concordance. - Fraction of TF ChIP-seq Peaks in Accessible Chromatin: Calculate the percentage of ChIP-seq peak summits falling within ATAC-seq or DNase-seq peaks. High overlap (>70-80%) supports valid TF binding in open chromatin.
- Statistical Enrichment Test: Use a tool like
BEDTools fisherto perform an odds ratio test, determining if the overlap between ChIP-seq peaks and an accessibility peak set is greater than expected by chance given genomic background.
Protocol: Orthogonal Validation Experiment
- Objective: Validate a subset of novel TF binding sites from ChIP-seq.
- Materials: Same cell line or tissue used for ChIP-seq.
- Procedure:
- Perform ATAC-seq on the target cells (see detailed protocol below).
- Process existing or new DNase-seq data from a comparable sample (e.g., ENCODE project).
- Intersect the ChIP-seq peak coordinates (BED file) with the unified set of accessibility peaks (ATAC-seq ∪ DNase-seq) using
BEDTools intersect. - Peaks not overlapping accessible regions are flagged for further scrutiny (potential false positives or artifact regions).
- Validate a selection of overlapping and non-overlapping peaks by independent method (e.g., PCR for footprint or ChIP-qPCR).
Detailed Experimental Protocol: ATAC-seq
Reagents: Cell permeabilization buffer (IGEPAL, Digitonin), Tagmentation buffer, Tn5 transposase (commercial kit, e.g., Illumina Tagment DNA TDE1), DNA purification beads (SPRI), PCR reagents, dual-indexed primers. Protocol:
- Harvest & Lysis: Pellet 50,000 viable cells. Wash with cold PBS. Resuspend in cold lysis buffer (10mM Tris-HCl pH7.4, 10mM NaCl, 3mM MgCl2, 0.1% IGEPAL, 0.1% Digitonin) for 3 min on ice. Immediately proceed.
- Tagmentation: Pellet nuclei, resuspend in transposase reaction mix (25 μL 2x TD Buffer, 2.5 μL TDE1, 22.5 μL nuclease-free water). Incubate at 37°C for 30 min in a thermomixer.
- DNA Purification: Immediately purify tagmented DNA using SPRI beads. Elute in 20 μL EB buffer.
- PCR Amplification: Amplify with indexed primers (Nextera index kits) for 10-12 cycles. Purify final library with SPRI beads.
- QC & Sequencing: Assess library profile (TapeStation/Fragment Analyzer; ~200-1000 bp smear). Sequence on Illumina platform (2x50 bp or 2x75 bp, minimum 25M reads for mammalian genomes).
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Integration Experiments
| Item | Function | Example/Product |
|---|---|---|
| Hyperactive Tn5 Transposase | Fragments and tags accessible chromatin for ATAC-seq. | Illumina Tagment DNA TDE1 / Enzyme Mix (Vazyme) |
| DNase I, RNase-free | Cleaves accessible DNA for DNase-seq. | Worthington DNase I (LS002139) |
| Cell Permeabilization Reagent | Lyses cell membrane while keeping nuclei intact for ATAC. | Digitonin (e.g., Millipore) |
| Dual-Indexed PCR Primers | Adds unique barcodes for multiplexed sequencing. | Illumina Nextera XT Index Kit v2 |
| SPRI Beads | Size-selective purification of DNA fragments post-tagmentation/PCR. | Beckman Coulter AMPure XP |
| High-Sensitivity DNA Assay | Quantifies low-concentration sequencing libraries. | Qubit dsDNA HS Assay Kit |
| Fragment Analyzer | Assesses library size distribution and quality. | Agilent 4200 TapeStation / Fragment Analyzer |
| Peak Calling Software | Identifies statistically significant enriched regions. | MACS2, F-Seq |
| Genomic Analysis Toolkit | Intersects, merges, and compares BED/GFF files. | BEDTools |
Visualizing the Integration Workflow
Cross-platform Validation Workflow for TF ChIP-seq
ATAC-seq vs DNase-seq Core Principles
Integrating ATAC-seq and DNase-seq data provides a robust framework for validating ChIP-seq findings in transcription factor research. This cross-platform approach mitigates platform-specific biases, increases confidence in identified regulatory elements, and refines the set of high-quality binding sites for downstream functional assays and drug target prioritization. Consistent application of the methodologies and analyses described herein will enhance the reproducibility and translational impact of chromatin profiling studies.
Comparative analysis of transcription factor (TF) binding is a critical step within the broader ChIP-seq data analysis workflow for transcription factor research. This analysis identifies genomic regions where TF occupancy significantly changes between biological conditions—such as disease versus healthy, treated versus untreated, or different cellular states. These differential binding events are pivotal for understanding transcriptional regulatory mechanisms driving phenotypic outcomes, with direct implications for target discovery in drug development.
Core Methodological Framework
The process integrates bioinformatics and statistical modeling to compare binding landscapes from multiple ChIP-seq experiments.
Experimental Design & Data Acquisition
- Biological Replicates: A minimum of two, but preferably three or more, independent biological replicates per condition are essential for statistical rigor.
- Controls: Appropriate controls (e.g., Input DNA, IgG, or non-specific antibody) must be sequenced for each condition to account for background noise and genomic biases.
- Sequencing Depth: Current standards recommend 20-50 million non-redundant, high-quality mapped reads per sample for mammalian genomes to ensure sufficient coverage for peak calling and comparison.
Computational & Statistical Analysis Pipeline
Primary Data Processing:
- Read Alignment: Map sequenced reads to a reference genome (e.g., using BWA, Bowtie2).
- Peak Calling: Identify significant binding sites (peaks) for each sample replicate independently (e.g., using MACS2, SPP).
- Peak Consistency: Generate a high-confidence set of peaks for each condition using irreproducible discovery rate (IDR) analysis or by merging replicates.
Differential Binding Analysis: This is performed using count-based models. Reads are counted in defined genomic intervals (consensus peak set) and analyzed with statistical tools designed for high-throughput sequencing data.
Table 1: Key Software Tools for Differential TF Binding Analysis
| Tool Name | Core Statistical Method | Key Feature | Best Use Case |
|---|---|---|---|
| DESeq2 | Negative Binomial Generalized Linear Model (GLM) | Robust dispersion estimation, handles complex designs. | Standard, well-replicated experiments. |
| edgeR | Negative Binomial GLM | Precise, good with low replication. | Experiments with limited replicates. |
| DiffBind | Wrapper for DESeq2/edgeR | Streamlined workflow from BAM files to results. | User-friendly integrated analysis. |
| ChIPComp | Beta-binomial model | Specifically incorporates background control data. | When matched Input controls are critical. |
Table 2: Quantitative Metrics for Interpreting Results
| Metric | Definition | Typical Significance Threshold |
|---|---|---|
| Log2 Fold Change (LFC) | Log2-transformed ratio of binding signal between conditions. | Absolute value > 1 (2-fold change) |
| False Discovery Rate (FDR)/Adjusted p-value | Probability that a called differential binding event is a false positive. | < 0.05 or < 0.01 |
| Read Counts (RPKM/CPM) | Reads Per Kilobase per Million or Counts Per Million; normalized signal intensity. | Used for visualization & filtering |
Detailed Protocol for Differential Binding Analysis Using DiffBind
This protocol assumes aligned BAM files and peak files (.narrowPeak or .bed) are available for all samples and replicates.
1. Create a Sample Sheet: Generate a comma-separated (.csv) file with columns: SampleID, Tissue, Factor, Condition, Treatment, Replicate, bamReads, ControlID, bamControl, Peaks, PeakCaller.
2. Read Data and Create a DBA Object:
3. Calculate Occupancy (Peak) Overlaps and Affinity (Read Count) Matrices:
4. Establish Contrast and Perform Differential Analysis:
5. Retrieve and Interpret Results:
6. Visualization: Generate MA plots, volcano plots, and heatmaps of binding affinities for differential sites.
Visualizing the Workflow
Title: Differential TF Binding Analysis ChIP-seq Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Comparative ChIP-seq Studies
| Item | Function & Rationale |
|---|---|
| High-Quality, Validated Antibodies | Specificity is paramount. Antibodies must be validated for ChIP (ChIP-grade) to ensure enrichment of the target TF with minimal cross-reactivity. |
| Chromatin Shearing Reagents | Consistent shearing to 200-500 bp fragments is critical. Uses sonication (e.g., Covaris shearing systems) or enzymatic (e.g., MNase, Tagmentase) methods. |
| Magnetic Protein A/G Beads | For efficient antibody-chromatin complex immunoprecipitation. Magnetic separation minimizes background. |
| Library Preparation Kits | Optimized for low-input and high-GC content DNA common in ChIP eluates (e.g., NEB Next Ultra II, SMARTer ThruPLEX). |
| Unique Dual-Indexed Sequencing Adapters | Enable multiplexing of many samples in one sequencing run, reducing batch effects and cost. Essential for cohort studies. |
| Spike-in Controls (e.g., D. melanogaster chromatin, S. pombe cells) | Added to samples before IP to normalize for technical variation (e.g., IP efficiency) between conditions, improving quantitative comparison. |
| Cell Line Authentication Kit | Confirms cell line identity using STR profiling, preventing misidentification that invalidates comparative results. |
| Viability/Cell Counting Assay | Ensures equal numbers of viable cells are used per IP across conditions, a key normalization factor. |
In the comprehensive ChIP-seq data analysis workflow for transcription factor research, raw data processing and peak calling are only the first steps. The critical phase of biological interpretation and validation relies heavily on integrating high-quality public reference data. The Encyclopedia of DNA Elements (ENCODE) and the Gene Expression Omnibus (GEO) serve as foundational resources for contextualizing novel findings, benchmarking analytical pipelines, and generating robust, testable hypotheses. This guide details a technical framework for their systematic use.
The Role of Public Data in the ChIP-seq Workflow
Following peak annotation and motif analysis, researchers must determine if their identified transcription factor binding sites (TFBS) are novel, tissue-specific, or part of a known regulatory program. ENCODE provides uniformly processed, gold-standard datasets for hundreds of transcription factors across numerous cell lines. GEO offers a vast repository of user-submitted data, enabling validation across diverse experimental conditions. Their integration answers key questions: Is the binding profile consistent with known biology? Does it correlate with histone marks or open chromatin in the same system? Are similar patterns observed in related tissues or diseases?
Querying and Acquiring Reference Datasets
ENCODE Portal (https://www.encodeproject.org/)
The ENCODE portal is searchable by target (e.g., CTCF), biosample (e.g., K562), assay (e.g., ChIP-seq), and file type. For validation, prioritise "optimal" or "replicated" datasets with high-quality metrics.
Key ENCODE Metadata & Quality Metrics (Representative Examples):
| Metric | Ideal Threshold / Value | Purpose in Validation |
|---|---|---|
| NRF (Non-Redundant Fraction) | > 0.9 | Indicates low PCR duplication, high library complexity. |
| PBC1 (PCR Bottlenecking Coefficient 1) | > 0.9 | Measures library complexity; lower values suggest over-amplification. |
| Cross-Correlation (NSC/ RSC) | NSC > 1.05, RSC > 1 | Assesses signal-to-noise in ChIP-seq; validates experiment quality. |
| Total Reads | > 20 million (for mammalian TFs) | Ensures sufficient depth for binding site detection. |
| Peak Calls (IDR) | Use IDR-thresholded peaks | Provides a conservative, reproducible set of high-confidence binding sites. |
GEO Database (https://www.ncbi.nlm.nih.gov/geo/)
Use advanced search with MeSH terms (e.g., "CTCF ChIP-seq" AND "heart"). Review associated publications for experimental details. Download raw FASTQ files or processed peak files (BED/narrowPeak).
Experimental Protocol for In-Silico Validation
Objective: To validate a novel CTCF ChIP-seq dataset from primary cardiomyocytes using public data.
Methodology:
Dataset Curation:
- From ENCODE, download IDR-thresholded peak files (BED format) and signal tracks (bigWig) for CTCF in the left ventricle of heart (E095 from the Roadmap Epigenomics Project, part of ENCODE) and in relevant model cell lines (e.g., H1-hESC).
- From GEO, search
"CTCF ChIP-seq cardiomyopathy". Download processed peak files from study GSE130051 (example).
Comparative Peak Analysis:
Overlap Analysis: Use
bedtools intersectto compute the overlap between your peaks and reference peaks. A significant overlap (e.g., >30% non-promoter peaks) supports validity.Motif Recovery: Use
MEME-ChIPorHOMERto find motifs in your peaks. Confirm the primary motif matches the canonical CTCF motif (JASPAR MA0139.1).
Signal Correlation:
Use
deepTools2to compute correlation of genome-wide signal between your bigWig and ENCODE bigWig files across all promoters or a set of conserved regulatory elements.A high Pearson correlation (r > 0.7) indicates strong technical and biological concordance.
Functional Contextualization:
- Integrate with publicly available histone mark data (H3K27ac for enhancers, H3K4me3 for promoters) from the same or similar biosample in ENCODE/GEO. This places your TFBS within an active regulatory landscape.
- Perform GREAT or ChIP-Enrich analysis on peaks unique to your condition to identify disease-relevant biological pathways.
Title: Public Data Integration Workflow for ChIP-seq Validation
The Scientist's Toolkit: Research Reagent Solutions
| Item / Resource | Function in Validation Workflow |
|---|---|
| ENCODE Portal & API | Programmatic access to download metadata and files using precise search terms (target, biosample, assay). |
| SRA Toolkit (NCBI) | Extracts FASTQ files from SRA archives (GEO's raw data storage) for re-analysis. |
| BEDTools Suite | Performs genomic arithmetic (intersect, merge, coverage) to compare peak sets quantitatively. |
| deepTools2 | Generates signal correlation matrices and aggregate plots (e.g., average profiles over TSS). |
| UCSC Genome Browser | Visualization hub for overlaying custom tracks with ENCODE reference tracks for visual inspection. |
| HOMER Suite | De novo motif discovery and enrichment analysis; verifies recovered motifs match known TF motifs. |
| GREAT or ChIP-Enrich | Assigns biological meaning to peak sets by linking genomic regions to downstream target genes and pathways. |
Title: Contextualizing TF Binding with Public Epigenomic Data
Table 1: Comparison of ENCODE and GEO for ChIP-seq Validation
| Feature | ENCODE | Gene Expression Omnibus (GEO) |
|---|---|---|
| Primary Use | Gold-standard reference; benchmarking. | Discovery; validation across diverse conditions. |
| Data Curation | Uniform processing pipelines, stringent QC. | Heterogeneous; user-submitted processing. |
| Metadata | Standardized, deep biosample annotation. | Variable; dependent on submitter. |
| Assay Breadth | Core set of TFs, histone marks, chromatin assays. | Unlimited; any published high-throughput data. |
| Ideal For | Technical quality control, defining consensus sites. | Biological context, disease mechanisms, novel systems. |
| Access Method | Portal, REST API. | Web search, SRA Toolkit. |
Table 2: Example ENCODE Metrics for CTCF ChIP-seq (K562 Cell Line)
| File Accession | Biosample | Total Reads | NRF | NSC (CC) | RSC (CC) | IDR Peaks | Purpose in Validation |
|---|---|---|---|---|---|---|---|
| ENCFF000OAZ | K562 | 45.2M | 0.97 | 1.52 | 1.21 | 91,452 | Primary reference for signal correlation. |
| ENCFF000OBE | K562 | 39.8M | 0.95 | 1.48 | 1.15 | 89,753 | Replicate for assessing reproducibility. |
Integrating ENCODE and GEO data transforms an isolated ChIP-seq result into a contextualized, biologically validated finding. This workflow ensures that subsequent functional experiments in transcription factor research are grounded in a solid comparative framework, accelerating the path from genomic observation to mechanistic insight and therapeutic discovery.
In a comprehensive thesis on ChIP-seq data analysis for transcription factor (TF) research, identifying genomic binding sites (peaks) is merely the first step. The pivotal biological question is: What are the functional consequences of this TF binding? Functional enrichment analysis translates a list of target genes, derived from ChIP-seq peaks, into interpretable biological knowledge. By statistically evaluating the over-representation of gene ontology (GO) terms or KEGG pathways, researchers can infer the TF's primary regulatory roles, implicated signaling cascades, and potential downstream phenotypic effects. This guide details the technical execution and interpretation of these analyses.
Gene Ontology (GO): A structured, controlled vocabulary describing gene functions across three domains:
- Biological Process (BP): Larger biological objectives (e.g., "inflammatory response").
- Molecular Function (MF): Molecular-level activities (e.g., "transcription factor binding").
- Cellular Component (CC): Locations within the cell (e.g., "nuclear chromatin").
KEGG Pathway Database: A collection of manually drawn pathway maps representing molecular interaction and reaction networks for metabolism, cellular processes, and human diseases.
Statistical Foundation: Hypergeometric test or Fisher's exact test is commonly used to assess whether the overlap between the submitted gene list and a given GO term/pathway is greater than expected by chance. P-values are adjusted for multiple testing (e.g., Benjamini-Hochberg FDR).
Current Data Sources (as of latest search): Analysis typically interfaces with consortium databases via R/Bioconductor packages (clusterProfiler, topGO) or web tools (DAVID, g:Profiler). These tools query updated versions of GO (released ~monthly) and KEGG (quarterly releases).
Experimental Protocol: From ChIP-seq Peaks to Enrichment
Input: A BED file of high-confidence ChIP-seq peaks for your transcription factor.
Step 1: Peak Annotation
- Tool:
ChIPseeker(R) orHOMER annotatePeaks.pl. - Method: Map each peak to the transcriptional start site (TSS) of the nearest gene, using a defined genomic window (e.g., -3kb to +1kb from TSS). Consider gene-based, not transcript-based, annotation.
- Output: A list of unique, putative target gene identifiers (e.g., Ensembl Gene IDs).
Step 2: Background Definition
- Critical Consideration: The background set must represent all genes that could have been detected in the experiment. This is typically all genes expressed in the cell type or, conservatively, all genes in the genome annotation. Never use only the significantly differentially expressed genes as the background for ChIP-seq-derived lists.
Step 3: Enrichment Analysis Execution (R/Bioconductor Example)
Step 4: Results Interpretation & Visualization
- Primary Output: Tables of enriched terms with p-values, adjustment q-values, and gene ratios.
- Visualization: Generate dotplots, barplots, enrichment maps, or cnetplots to show relationships between genes and terms.
- Downstream Integration: Correlate enriched processes with phenotypic data from TF perturbation experiments (e.g., knockout RNA-seq).
Quantitative Data Presentation
Table 1: Top Enriched GO Biological Processes in a Hypothetical TF ChIP-seq Study
| GO Term ID | Description | Gene Ratio (Target/Background) | P-value | Adjusted Q-value | Target Gene Count |
|---|---|---|---|---|---|
| GO:0045944 | Positive regulation of transcription by RNA polymerase II | 45/1200 (0.038) | 1.2e-12 | 3.5e-09 | 45 |
| GO:0000122 | Negative regulation of transcription by RNA polymerase II | 32/1200 (0.027) | 5.8e-08 | 8.2e-05 | 32 |
| GO:0006366 | Transcription by RNA polymerase II | 58/1200 (0.048) | 2.1e-07 | 0.00021 | 58 |
| GO:0045893 | Positive regulation of DNA-templated transcription | 48/1200 (0.040) | 3.4e-07 | 0.00025 | 48 |
Table 2: Top Enriched KEGG Pathways from the Same Analysis
| Pathway ID | Pathway Name | Gene Ratio (Target/Background) | P-value | Adjusted Q-value | Target Gene Count |
|---|---|---|---|---|---|
| hsa04010 | MAPK signaling pathway | 28/1200 (0.023) | 4.5e-06 | 0.0032 | 28 |
| hsa04310 | Wnt signaling pathway | 18/1200 (0.015) | 1.1e-04 | 0.039 | 18 |
| hsa05205 | Proteoglycans in cancer | 22/1200 (0.018) | 0.00015 | 0.042 | 22 |
| hsa04151 | PI3K-Akt signaling pathway | 25/1200 (0.021) | 0.00032 | 0.057 | 25 |
Pathway and Workflow Visualizations
Title: ChIP-seq Functional Enrichment Analysis Workflow
Title: Simplified MAPK Signaling Pathway (KEGG hsa04010)
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for ChIP-seq & Downstream Functional Analysis
| Item | Function in Workflow | Example/Note |
|---|---|---|
| TF-Specific Antibody | Immunoprecipitation of the transcription factor-DNA complex. | High specificity validated for ChIP is critical (e.g., Diagenode, Cell Signaling). |
| Protein A/G Magnetic Beads | Capture of antibody-bound complexes. | Efficient for washing and reducing background. |
| Crosslinking Reagent | Formaldehyde fixes protein-DNA interactions. | Typically 1% final concentration. |
| Chromatin Shearing Kit | Fragment chromatin to 200-600 bp via sonication. | Includes optimized buffers and protocols (e.g., Covaris, Bioruptor). |
| High-Fidelity DNA Library Prep Kit | Prepares ChIP DNA for next-generation sequencing. | Must handle low-input DNA (e.g., Illumina, NEB Next). |
| Genome Annotation Package (e.g., TxDb.Hsapiens.UCSC.hg38.knownGene) | Provides gene model coordinates for peak annotation. | Bioconductor package corresponding to your reference genome. |
| Functional Enrichment Software | Performs statistical over-representation analysis. | R: clusterProfiler, topGO; Web: g:Profiler, Enrichr. |
| Pathway Visualization Tool | Generates custom pathway diagrams. | Cytoscape, Pathview (R), KEGG Mapper. |
The systematic analysis of Transcription Factor (TF) binding landscapes via ChIP-seq has evolved from a basic discovery tool to a cornerstone of mechanistic and translational biology. Within a comprehensive ChIP-seq data analysis workflow, the critical translational step involves mapping TF-bound cis-regulatory elements (CREs) to target genes and intersecting these networks with human genetic data. This guide details the protocols and analytical frameworks required to move from peak calling to clinically actionable insights, focusing on how aberrant TF binding drives disease pathogenesis and presents opportunities for therapeutic intervention.
Foundational ChIP-seq Workflow for TF Analysis
A robust ChIP-seq pipeline is prerequisite for any downstream translational application.
Experimental Protocol: Core ChIP-seq for Transcription Factors
- Cell/Tissue Fixation: Crosslink proteins to DNA using 1% formaldehyde for 10 min at room temperature. Quench with 125mM glycine.
- Cell Lysis & Chromatin Shearing: Lyse cells in SDS lysis buffer. Shear chromatin to 200-500 bp fragments via optimized sonication (e.g., Bioruptor, 8 cycles of 30 sec ON/30 sec OFF). Immunoprecipitate with 2-5 µg of validated, target-specific TF antibody.
- Immunoprecipitation & Washing: Incubate sheared chromatin with antibody-bound beads overnight at 4°C. Wash sequentially with Low Salt, High Salt, LiCl, and TE buffers.
- Reverse Crosslinking & Purification: Reverse crosslinks at 65°C overnight with proteinase K. Recover DNA via phenol-chloroform extraction and ethanol precipitation.
- Library Preparation & Sequencing: Use a kit (e.g., NEBNext Ultra II DNA) for end-repair, dA-tailing, adapter ligation, and PCR enrichment. Sequence on an Illumina platform to a depth of 20-40 million reads.
Data Analysis Workflow Summary:
- Quality Control & Alignment: FastQC, Trim Galore, alignment with Bowtie2/BWA.
- Peak Calling: MACS2 for broad or narrow peaks (--broad for histone marks, default for TFs).
- Downstream Analysis: motif discovery (HOMER, MEME-ChIP), annotation (ChIPseeker), integrative analysis.
Translational Analytical Framework: Linking Peaks to Disease
The key translational step is integrating TF binding data with orthogonal genomic and clinical datasets.
Table 1: Key Integrative Datasets for Translational TF Research
| Dataset Type | Primary Source | Key Translational Application |
|---|---|---|
| Genome-Wide Association Studies (GWAS) | NHGRI-EBI GWAS Catalog | Colocalization of TF binding sites with disease-associated non-coding SNPs. |
| Quantitative Trait Loci (QTLs) | GTEx, eQTL Catalogue | Linking TF-bound CREs to gene expression regulation in disease-relevant tissues. |
| Somatic Mutations in Cancer | COSMIC, TCGA | Identifying non-coding mutations disrupting or creating de novo TF binding motifs. |
| Chromatin Accessibility | ENCODE, Roadmap Epigenomics | Defining cell-type-specific active regulatory landscapes for TF binding context. |
Protocol: Integrative Analysis of TF Binding with GWAS SNPs
- Input: High-confidence TF peaks (BED file) and GWAS SNP coordinates (GWAS Catalog).
- Tool: Use
BEDTools intersector specialized tools likeGARFIELD. - Method:
- Liftover GWAS SNP coordinates (hg38) if necessary.
- Intersect SNP coordinates with TF peak regions (±50-100 bp).
- Annotate overlapping SNPs to the nearest gene(s) and regulatory domain (using TAD maps).
- Perform in silico motif analysis (
FIMO,HOMER) to assess if the SNP alters the predicted binding affinity for the TF or co-factors. - Validate candidate SNP effects via reporter assays (e.g., luciferase) in relevant cell lines.
Clinical Applications and Disease Case Studies
Oncology: TP53 Mutations and Altered Cistromes Mutant p53 exhibits oncogenic gain-of-function by binding novel genomic loci, activating pro-proliferative genes.
- Experiment: Perform ChIP-seq for mutant p53 (R175H, R273H) vs. wild-type in isogenic cell lines.
- Analysis: Identify "neo-binding sites," correlate with open chromatin (ATAC-seq), and link to upregulated oncogenic pathways (e.g., KRAS signaling).
Autoimmune Disease: NF-κB in Rheumatoid Arthritis Constitutive NF-κB activation in synovial fibroblasts drives chronic inflammation.
- Experiment: ChIP-seq for p65/RelA in patient-derived synovial fibroblasts stimulated with TNF-α.
- Translation: Intersect peaks with RA-risk SNPs. Identify super-enhancers co-occupied by NF-κB and lineage-determining TFs, revealing master regulators of disease phenotype.
Neurodegeneration: MEF2 in Alzheimer's Disease Oxidative stress in neurons leads to loss of neuroprotective MEF2 binding.
- Experiment: ChIP-seq for MEF2 in neuronal cell models under oxidative stress.
- Translation: Identify lost binding events at promoters of synaptic integrity genes. Correlate with epigenetic silencing marks (H3K27me3 ChIP-seq).
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents for Translational TF Studies
| Reagent / Material | Function & Application |
|---|---|
| Validated ChIP-Grade Antibodies (e.g., Diagenode, Cell Signaling) | High-specificity antibodies for the target TF, essential for clean ChIP-seq signal. |
| Magna ChIP Protein A/G Beads (MilliporeSigma) | Magnetic beads for efficient antibody-chromatin complex pulldown and low-background washes. |
| NEBNext Ultra II DNA Library Prep Kit (NEB) | Robust, high-yield library preparation from low-input ChIP DNA. |
| Tn5 Transposase (Tagmentase) | For simultaneous fragmentation and tagging in ATAC-seq, mapping open chromatin complementary to TF binding. |
| CRISPR/dCas9-KRAB or dCas9-p300 Systems | For functional validation: repress or activate candidate CREs to test gene regulation and phenotypic impact. |
| Luciferase Reporter Vectors (pGL4-series, Promega) | Validate the regulatory activity of TF-bound CREs and the functional impact of disease-associated SNPs. |
| Patient-Derived Primary Cells or iPSCs | Disease-relevant cellular models for translational studies, preserving genetic and epigenetic context. |
Pathway and Workflow Visualizations
Title: Translational TF ChIP-seq Analysis Workflow
Title: NF-κB Pathway Dysregulation by Risk SNP
Therapeutic Implications and Future Directions
Mapping disease-critical TF binding sites directly informs therapeutic development.
- Small Molecules: Targeting pathological TF activity (e.g., BET inhibitors for MYC).
- Gene Therapy: CRISPR-based editing of aberrant CREs.
- Biomarkers: Circulating nucleosomes from TF-bound regions as non-invasive biomarkers.
The integration of high-quality TF binding maps with human genetics is an indispensable strategy for deconvoluting the regulatory logic of disease, bridging the gap between non-coding genetic variation and mechanistic pathophysiology.
Best Practices for Reproducibility and Data Sharing in Publications
In the context of a specialized workflow for ChIP-seq data analysis in transcription factor research, implementing robust reproducibility and data sharing practices is non-negotiable. This technical guide outlines the foundational principles and actionable steps required to ensure that computational biology research can be independently verified and built upon.
Foundational Principles and Quantitative Benchmarks
Adherence to the FAIR (Findable, Accessible, Interoperable, Reusable) data principles is the cornerstone of modern reproducible research. Quantitative studies highlight the persistent gaps and the impact of proper practices.
Table 1: Current State and Impact of Reproducibility Practices in Genomics
| Metric | Reported Value (%) / Number | Source / Year | Implication for ChIP-seq Workflows |
|---|---|---|---|
| Studies providing public data availability | ~70% (GEO/SRA) | NIH Genomic Data Sharing Policy, 2024 | Mandatory for most funded research; private during peer review is standard. |
| Studies with fully executable code | <30% | Review of 2023 bioRxiv preprints | Major barrier to replicating peak calling, motif analysis, and differential binding. |
| Reproducibility rate of published results | 50-80% (varies by sub-field) | Various meta-analyses, 2020-2024 | Underlines critical need for detailed workflow and parameter documentation. |
| Citation advantage for shared data | +25% to +50% | Piwowar et al., 2013; subsequent confirmations | Strong incentive for depositing raw FASTQ and processed bigWig/BED files. |
Detailed Methodologies for a Reproducible ChIP-seq Workflow
A replicable ChIP-seq analysis for transcription factors depends on meticulous documentation at every stage.
Experimental Protocol: Wet-Lab ChIP-seq for a Transcription Factor
- Crosslinking: Treat cells with 1% formaldehyde for 10 minutes at room temperature. Quench with 125mM glycine.
- Sonication: Lyse cells and shear chromatin via sonication (e.g., Covaris S220, 200 cycles/burst, 60 sec duty cycle) to achieve 200-500 bp fragments. Verify size on agarose gel.
- Immunoprecipitation: Incubate clarified lysate with validated, target-specific antibody (e.g., 2-5 µg) overnight at 4°C. Use Protein A/G beads for capture.
- Wash & Elution: Wash beads stringently (e.g., low salt, high salt, LiCl, TE buffers). Elute complexes with fresh elution buffer (1% SDS, 100mM NaHCO3).
- Reverse Crosslinks & Purification: Incubate eluate at 65°C overnight with NaCl. Treat with RNase A and Proteinase K. Purify DNA using silica columns.
- Library Prep & Sequencing: Use a standardized library preparation kit (e.g., Illumina TruSeq). Sequence on an appropriate platform (e.g., Illumina NovaSeq) to obtain a minimum of 20 million paired-end 50bp reads per sample.
Computational Protocol: Analysis Workflow from RAW Data to Peaks
- Data and Tool Availability: All raw sequencing files (FASTQ) must be deposited in a public repository like the Sequence Read Archive (SRA) or GEO. All analysis code must be shared on a version-controlled platform like GitHub or GitLab.
- Quality Control: Use FastQC v0.12.1 for initial quality assessment. Trim adapters and low-quality bases using Trim Galore! v0.6.10 (wrapper for Cutadapt and FastQC).
- Alignment: Align reads to the appropriate reference genome (e.g., GRCh38/hg38) using a splice-aware aligner like BWA-MEM v0.7.17 or Bowtie2 v2.5.1. Filter for uniquely mapped, properly paired reads.
- Post-Alignment Processing: Remove PCR duplicates using Picard MarkDuplicates v2.27.5. Generate coverage tracks (bigWig files) using deepTools bamCoverage v3.5.3, normalized to Reads Per Genome Coverage (RPGC).
- Peak Calling: Call peaks for the transcription factor sample against its matched input/control using MACS2 v2.2.7.1 with a q-value (FDR) cutoff of 0.05. Crucially, document all parameters:
--call-summits,--shift,--extsize, etc. - Downstream Analysis: Perform motif discovery on summit sequences (±100bp) using HOMER v4.11 or MEME-ChIP. Integrate with RNA-seq or ATAC-seq data for functional context using tools like bedtools.
Visualization of Workflows
ChIP-seq Analysis & Sharing Workflow
FAIR Principles to Reproducibility Pipeline
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools and Reagents for Reproducible ChIP-seq Research
| Item | Function in Workflow | Example/Standard | Critical for Reproducibility |
|---|---|---|---|
| Validated Antibody | Specific immunoprecipitation of target transcription factor. | Commercial (CST, Abcam) with cited ChIP-grade validation. | Provide catalog #, lot #, RRID. Negative control antibody essential. |
| Crosslinking Reagent | Fixes protein-DNA interactions. | Formaldehyde, 1% solution. | Specify vendor, concentration, incubation time. |
| Sonication System | Shears chromatin to optimal fragment size. | Covaris S220, Bioruptor Pico. | Document exact settings (Wattage, Cycles, Time). Provide gel image of sheared DNA. |
| Sequencing Platform | Generates raw sequencing reads. | Illumina NovaSeq, NextSeq. | State platform, read length (e.g., 2x50bp), and minimum depth (e.g., 20M reads). |
| Reference Genome | Alignment and annotation baseline. | UCSC hg38, ENSEMBL GRCh38. | Specify exact version and source (e.g., GENCODE v44). |
| Analysis Pipeline | Standardized processing and peak calling. | nf-core/chipseq, PEPATAC. | Using a versioned, containerized pipeline (Docker/Singularity) ensures computational replicability. |
| Data Repository | Public archiving of raw and processed data. | GEO, SRA, ENCODE portal. | Mandatory for publication. Use structured metadata templates. |
| Code Repository | Version control and sharing of analysis code. | GitHub, GitLab, Zenodo (for snapshots). | Include a detailed README, environment file (conda.yml), and run scripts. |
Conclusion
Successful ChIP-seq analysis for transcription factors requires careful integration of experimental design, computational methodology, troubleshooting expertise, and rigorous validation. By following this comprehensive workflow, researchers can reliably identify TF binding events, understand their functional implications, and generate biologically meaningful insights. The convergence of improved antibodies, higher sequencing depth, and advanced analytical tools continues to enhance our ability to decode transcriptional regulation. Future directions include single-cell ChIP-seq applications, integration with multi-omics datasets, and the development of machine learning approaches to predict TF binding dynamics. These advances will further empower drug development professionals to identify novel therapeutic targets by precisely mapping the regulatory landscape in both normal physiology and disease states.