Navigating the Multi-omics Universe: A 2024 Guide to Essential Data Repositories and Research Resources
This guide provides researchers, scientists, and drug development professionals with a comprehensive overview of the multi-omics data landscape.
Navigating the Multi-omics Universe: A 2024 Guide to Essential Data Repositories and Research Resources
Abstract
This guide provides researchers, scientists, and drug development professionals with a comprehensive overview of the multi-omics data landscape. It covers foundational public repositories, practical methodologies for accessing and integrating diverse data types, strategies to overcome common technical and analytical challenges, and best practices for validating data quality and comparing resource utility. The article synthesizes current resources to empower efficient, reproducible, and translatable multi-omics research.
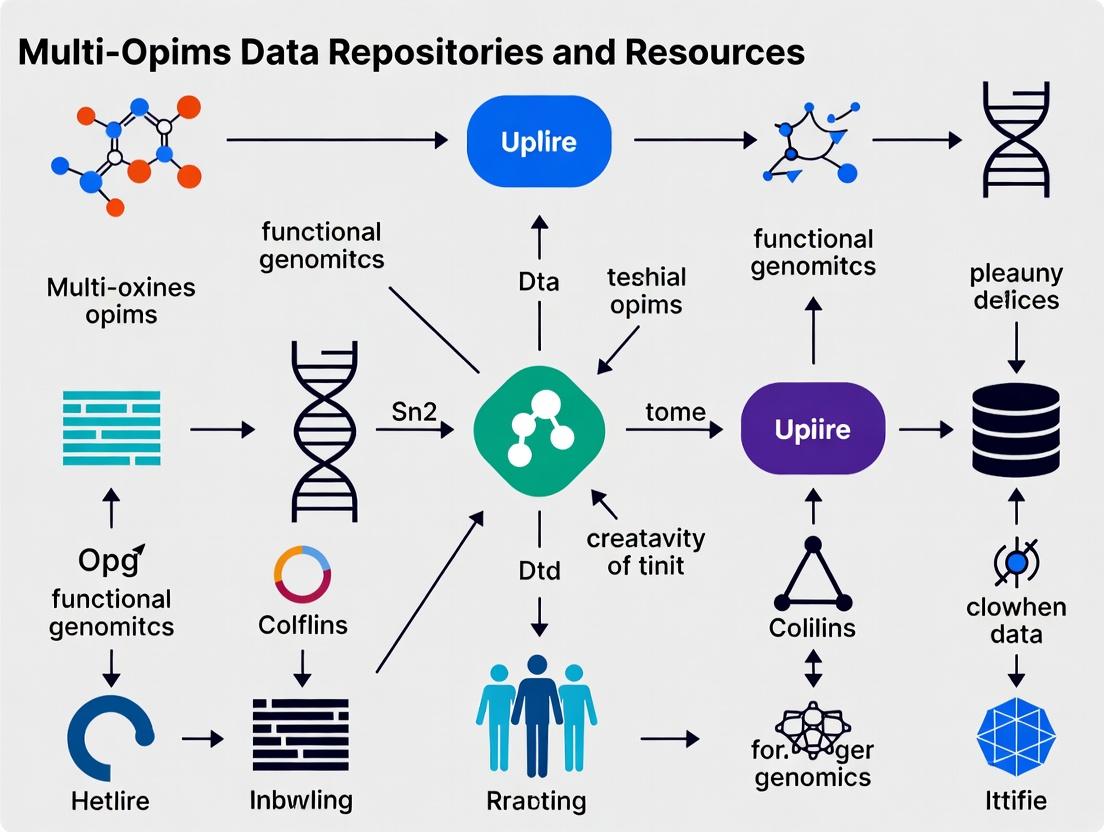
The Multi-omics Landscape: Discovering Core Public Repositories and Data Portals
Within the context of advancing multi-omics data repositories and resources, a systematic understanding of the core "omics" disciplines is foundational. This technical guide details the hierarchy, methodologies, and integration points of the modern omics stack, which forms the bedrock of systems biology and precision medicine initiatives.
The Omics Hierarchy: From DNA to Phenotype
The central dogma of molecular biology provides the conceptual framework for the omics stack, each layer capturing a distinct level of biological information. The sequential and regulatory relationships between these layers are complex and non-linear.
Title: Hierarchical Flow of Information in the Omics Stack
Core Omics Disciplines: Quantitative Scope & Key Technologies
Each layer of the omics stack is characterized by its unique molecular entities, scale, and the dominant high-throughput technologies used for its interrogation.
| Omics Layer | Primary Molecule | Approximate Scale in Humans | Dominant High-Throughput Technology | Key Repositories (Examples) |
|---|---|---|---|---|
| Genomics | DNA | ~3.2 billion base pairs (haploid) | Next-Generation Sequencing (NGS), Microarrays | dbSNP, gnomAD, dbGaP |
| Epigenomics | Chromatin, DNA/Histone Modifications | ~28 million CpG sites, numerous histone marks | Bisulfite-Seq, ChIP-Seq, ATAC-Seq | ENCODE, Roadmap Epigenomics |
| Transcriptomics | RNA (mRNA, ncRNA) | ~20,000 coding genes, >100,000 transcripts | RNA-Seq, Microarrays | GEO, SRA, GTEx |
| Proteomics | Proteins & Peptides | ~20,000 canonical proteins, >1 million proteoforms | Mass Spectrometry (LC-MS/MS), Antibody Arrays | PRIDE, ProteomeXchange |
| Metabolomics | Metabolites | ~10,000+ detectable metabolites | Mass Spectrometry (GC/LC-MS), NMR | Metabolights, HMDB |
Detailed Experimental Protocols
Bulk RNA-Sequencing (Transcriptomics)
Objective: To profile the abundance and sequence of RNA molecules in a biological sample.
Detailed Protocol:
- RNA Extraction & QC: Isolate total RNA using guanidinium thiocyanate-phenol-chloroform extraction (e.g., TRIzol). Assess purity (A260/A280 ~2.0) and integrity (RIN > 8.0) using a Bioanalyzer.
- Library Preparation:
- Poly-A Selection: Enrich mRNA using oligo(dT) beads.
- Fragmentation: Chemically or enzymatically fragment RNA to ~200-300bp.
- cDNA Synthesis: Perform first-strand synthesis using reverse transcriptase and random hexamers, followed by second-strand synthesis.
- End Repair, A-tailing & Adapter Ligation: Convert cDNA ends to blunt ends, add an 'A' overhang, and ligate sequencing adapters with unique dual indices (UDIs) for multiplexing.
- PCR Amplification: Enrich adapter-ligated fragments (typically 10-12 cycles).
- Sequencing: Pool libraries and sequence on an Illumina platform (e.g., NovaSeq) to a depth of 20-50 million paired-end reads per sample.
- Bioinformatics Pipeline: Use tools like FastQC for quality control, STAR for alignment to a reference genome, and featureCounts for gene-level quantification.
Shotgun Proteomics via LC-MS/MS
Objective: To identify and quantify proteins in a complex sample.
Detailed Protocol:
- Protein Extraction & Digestion: Lyse cells/tissues in a denaturing buffer (e.g., 8M Urea). Reduce disulfide bonds with DTT and alkylate with iodoacetamide. Digest proteins to peptides using trypsin (1:50 enzyme-to-substrate ratio, 37°C, overnight).
- Peptide Desalting: Use C18 solid-phase extraction (SPE) tips or stage tips to desalt and concentrate peptides.
- Liquid Chromatography (LC): Separate peptides on a reverse-phase C18 column (75µm x 25cm) using a nanoflow LC system with a gradient from 2% to 35% acetonitrile over 120 minutes.
- Mass Spectrometry (MS):
- Full Scan (MS1): Eluting peptides are ionized (ESI) and analyzed in the Orbitrap mass analyzer (resolution 120,000; scan range 350-1500 m/z).
- Data-Dependent Acquisition (DDA): The top 20 most intense precursor ions from MS1 are isolated, fragmented by HCD (collision energy 28%), and the fragment ions analyzed in the Orbitrap (resolution 15,000). Dynamic exclusion is set to 30 seconds.
- Data Analysis: Search MS/MS spectra against a protein sequence database (e.g., UniProt Human) using engines like MaxQuant or FragPipe, allowing for fixed carbamidomethylation and variable methionine oxidation modifications.
Multi-Omic Integration: A Conceptual Workflow
The power of the omics stack is realized through integration. A typical workflow for correlating data across genomic, transcriptomic, and proteomic layers to identify driver mechanisms is outlined below.
Title: Multi-Omic Data Integration & Validation Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Vendor Examples | Primary Function in Omics Experiments |
|---|---|---|
| TRIzol/ Qiazol | Thermo Fisher, Qiagen | Simultaneous isolation of RNA, DNA, and proteins from a single sample. Essential for matched multi-omic analysis. |
| DNase I (RNase-free) | New England Biolabs, Roche | Removal of contaminating genomic DNA from RNA preparations prior to RNA-Seq or qPCR. |
| Nextera XT DNA Library Prep Kit | Illumina | Rapid, tagmentation-based preparation of sequencing libraries from low-input DNA for genomics/epigenomics. |
| KAPA HyperPrep Kit | Roche | Robust library preparation for RNA-Seq, offering high complexity and uniformity. |
| Trypsin, Sequencing Grade | Promega, Thermo Fisher | Proteolytic enzyme for specific digestion of proteins at lysine and arginine residues for bottom-up proteomics. |
| TMTpro 16plex Isobaric Label Reagents | Thermo Fisher | Set of 16 isobaric chemical tags for multiplexed quantitative comparison of up to 16 proteome samples in a single MS run. |
| C18 StageTips | Thermo Fisher | Micro-columns for desalting and concentrating peptide samples prior to LC-MS/MS analysis. |
| Bioanalyzer High Sensitivity DNA/RNA Chips | Agilent Technologies | Microfluidics-based electrophoresis for precise assessment of nucleic acid fragment size distribution and integrity (RIN). |
Within the thesis framework of Multi-omics data repositories and resources research, the efficient discovery and retrieval of primary data is foundational. The National Center for Biotechnology Information (NCBI, USA), the European Bioinformatics Institute of the European Molecular Biology Laboratory (EBI-EMBL, Europe), and the DNA Data Bank of Japan (DDBJ) constitute the International Nucleotide Sequence Database Collaboration (INSDC). These NIH and internationally sponsored powerhouses are the universal, canonical starting points for genomic, transcriptomic, and epigenomic data. This guide details their core functions, access protocols, and integrative use in modern multi-omics workflows.
Core Repository Comparison
A live search confirms these repositories maintain synchronized primary nucleotide data, but their tools, additional databases, and user interfaces differ significantly.
Table 1: Quantitative Comparison of Core Resources (as of 2024)
| Feature | NCBI | EBI-EMBL (EBI-E) | DDBJ |
|---|---|---|---|
| Primary Portal | https://www.ncbi.nlm.nih.gov | https://www.ebi.ac.uk | https://www.ddbj.nig.ac.jp |
| Total Records (INSDC) | ~2.5 Petabases (shared across INSDC) | ~2.5 Petabases (shared across INSDC) | ~2.5 Petabases (shared across INSDC) |
| Key Unique Tools | BLAST, PubMed, dbSNP, ClinVar, SRA | UniProt, Ensembl, PRIDE, ArrayExpress, MGnify | DDBJ Search, JGA, NBDC Human Database |
| Omics Specialization | Genomics (SRA, dbGaP), Literature | Proteomics (PRIDE), Metagenomics (MGnify), Functional (Ensembl) | Asian Genomes, NGS (DRA), Human (JGA) |
| Programmatic Access | E-utilities API, Datasets API | REST APIs (e.g., UniProt, ENA), BioMart | DDBJ API, NBDC API |
| Submission Platform | Submission Portal (BankIt, tbl2asn) | Webin (ENA, PRIDE, MetaboLights) | DDBJ Submission System (NSSS, D-way) |
Table 2: Multi-Omics Data Type Mapping
| Data Type | NCBI Resource | EBI-EMBL Resource | DDBJ Resource |
|---|---|---|---|
| Genomics (Raw) | Sequence Read Archive (SRA) | European Nucleotide Archive (ENA) | DDBJ Sequence Read Archive (DRA) |
| Genomics (Variants) | dbSNP, dbVar | EVA (European Variation Archive) | JGA (for controlled-access) |
| Transcriptomics | GEO, SRA | ArrayExpress, ENA | DRA, GEO (mirrored) |
| Proteomics | (Limited - via Identical Protein) | PRIDE, UniProt | (Limited - via JGA) |
| Metabolomics | (Limited) | MetaboLights | (Limited) |
| Metagenomics | (via SRA) | MGnify | DRA |
Experimental Protocols for Data Retrieval & Integration
Protocol 1: Bulk Download of RNA-Seq Data from a GEO/SRA Study Objective: Programmatically retrieve raw sequencing files (FASTQ) for a defined set of samples.
- Identify Accession: Locate the Series accession (e.g., GSE123456) on NCBI GEO or the Study accession (e.g., SRP123456) on SRA/EBI-EMBL's ENA.
- Fetch Metadata: Use NCBI's
efetch(E-utilities) or ENA's REST API to obtain sample-level metadata, linking experiment (SRX) to run (SRR) accessions.- NCBI Command (E-utilities):
esearch -db sra -query "SRP123456" | efetch -format runinfo > metadata.csv - EBI-EMBL Command (curl):
curl "https://www.ebi.ac.uk/ena/portal/api/filereport?accession=SRP123456&result=read_run&fields=run_accession,fastq_ftp" > ftp_links.txt
- NCBI Command (E-utilities):
- Generate Download Script: Parse the metadata to create a shell script with
wgetoraspera(ascp) commands for eachfastq_ftplink. - Integrate with Analysis Pipeline: Directly pass the downloaded file paths to a workflow manager (Nextflow, Snakemake) for quality control (FastQC), alignment (HISAT2, STAR), and quantification (featureCounts, Salmon).
Protocol 2: Cross-Referencing a Genetic Variant to Functional Annotation Objective: From a dbSNP (NCBI) variant ID, obtain population frequency, clinical significance, and genomic context.
- Variant Lookup: Query
rs123456via NCBI's Variation Viewer or thesnpdatabase usingefetch. - Retrieve Linked Data: Extract genomic coordinates (chr, pos), allele frequencies from gnomAD (broadly available via Ensembl/EBI), and clinical assertions from ClinVar (NCBI).
- Lift-Over to Functional Genome Browser: Use the genomic coordinates to view the variant in its genomic context via EBI-EMBL's Ensembl genome browser. This provides data on overlapping genes, regulatory elements, and conserved regions.
- Pathway Contextualization: If the variant lies within a protein-coding gene, use the linked UniProt (EBI-EMBL) entry to identify the protein's role in signaling pathways (e.g., via Reactome).
Visualizing the Multi-Omics Data Integration Workflow
Title: Data flow between INSDC repositories and researcher analysis.
Title: Cross-referencing a variant from NCBI to EBI resources.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Digital Research Reagents for Multi-Omics Discovery
| Item (Tool/Resource) | Primary Source | Function in Workflow |
|---|---|---|
| SRA Toolkit | NCBI | A suite of tools for downloading, converting, and manipulating data from the Sequence Read Archive (SRA). |
| E-utilities (Entrez Direct) | NCBI | Command-line tools for accessing NCBI databases programmatically, enabling automated queries and data pipeline integration. |
| ENA Browser & API | EBI-EMBL | Web interface and RESTful API for searching and retrieving data from the European Nucleotide Archive, including fastq files and metadata. |
| BioMart | EBI-EMBL | Data mining tool for complex queries across Ensembl genomes, facilitating bulk extraction of gene IDs, sequences, and annotations. |
| Aspera Client | IBM (used by INSDC) | High-speed file transfer client required for the fastest download of large sequencing datasets from SRA, ENA, or DRA. |
| DDBJ FTP Server Access | DDBJ | Reliable FTP-based bulk download site for publicly available DDBJ/DRA data, often integrated into batch scripts. |
| Galaxy Project Tools | Community (hosted by EBI/others) | Web-based platform providing accessible, reproducible workflows for multi-omics analysis, linking directly to repository data. |
Within the framework of multi-omics data repositories and resources research, integrating human disease data with model organism information is fundamental for translational discovery. This guide details the core functions, data structures, and integration methodologies for four pivotal resource hubs: The Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO), Sequence Read Archive (SRA), and Model Organism Databases (MODs). The convergence of these resources enables the identification of conserved molecular hubs across species, accelerating target validation and drug development.
Modern biomedical research relies on cross-species data integration. Human-centric repositories like TCGA, GEO, and SRA provide disease-specific molecular profiles, while MODs offer deep genetic, phenotypic, and experimental context for key species. Identifying orthologous genes and pathways that serve as functional "hubs" across these datasets is a powerful strategy for prioritizing therapeutic targets and understanding disease mechanisms.
Core Repository Specifications and Data Access
Table 1: Core Characteristics of Primary Data Hubs
| Repository | Primary Focus | Data Types | Key Access Tools/APIs | Typical Use Case in Hub Identification |
|---|---|---|---|---|
| The Cancer Genome Atlas (TCGA) | Human Cancer Genomics | WGS, WES, RNA-Seq, miRNA, Methylation, Clinical | GDC Data Portal, TCGAbiolinks (R), GDC API | Identifying differentially expressed and mutated genes in cancer vs. normal tissue. |
| Gene Expression Omnibus (GEO) | Functional Genomics | Microarray, RNA-Seq, SNP, Methylation, CHIP-Seq | GEOquery (R), SRAdb, Web Interface | Finding public gene expression signatures for diseases and treatments. |
| Sequence Read Archive (SRA) | Raw Sequencing Data | Raw reads (FASTQ), Alignment data | SRA Toolkit, SRAdb (R), E-utilities | Downloading raw data for custom re-analysis or novel integration. |
| Model Organism Databases (e.g., MGI, FlyBase, WormBase) | Model Organism Biology | Genomes, annotations, phenotypes, orthologs, pathways | Direct download, BioMart, species-specific APIs | Mapping human disease genes to orthologs and retrieving mutant phenotypes. |
| Resource | Estimated Datasets/Studies | Estimated Samples | Key Organisms | Update Frequency |
|---|---|---|---|---|
| TCGA (via GDC) | ~84 projects (e.g., TCGA-BRCA) | >11,000 patients (tumor/normal) | Homo sapiens | Finalized; maintained |
| GEO | >150,000 series | >5 million samples | All | Daily |
| SRA | >40 Petabases of data | Tens of millions of runs | All | Continuous |
| MGI (Mouse) | >73,000 genes annotated | Millions of mutant phenotypes | Mus musculus | Weekly |
| FlyBase | ~18,000 genes | ~290,000 alleles | Drosophila melanogaster | Daily/Weekly |
| WormBase | ~20,000 genes | ~175,000 variation alleles | Caenorhabditis elegans | Monthly |
Experimental Protocol: Identifying and Validating a Conserved Disease Hub
This protocol outlines a standard computational-experimental pipeline for identifying a gene/protein hub using these resources.
Objective: To identify a candidate oncogene from TCGA, analyze its expression signature in GEO, and validate its functional role using a model organism.
Phase 1: Computational Discovery from Human Data
TCGA Data Extraction:
- Access TCGA-BRCA RNA-Seq HTSeq counts and clinical data using the
TCGAbiolinksR package. - Perform differential expression analysis (
TCGAbiolinks::TCGAanalyze_DEA) between tumor (primary solid tumor) and normal (solid tissue normal) samples. Apply FDR correction (Benjamini-Hochberg). - Filter for genes with |log2FC| > 2 and FDR < 0.01.
- Perform survival analysis (
survivalpackage) using Kaplan-Meier plots for top upregulated genes.
- Access TCGA-BRCA RNA-Seq HTSeq counts and clinical data using the
Cross-Validation in GEO:
- Identify a relevant GEO series (e.g., GSE12345 for breast cancer drug response).
- Use
GEOqueryto download the series matrix and platform data. - Normalize and analyze differential expression (using
limmafor microarray) to confirm the candidate gene's association with the phenotype of interest.
Ortholog Mapping:
- Query the candidate human gene (e.g., EGFR) in the Alliance of Genome Resources or individual MODs (MGI, FlyBase) to retrieve high-confidence orthologs (e.g., Egfr in mouse, Egfr in fly).
- Retrieve known phenotypes, mutant alleles, and available reagents for the ortholog.
Phase 2: Experimental Validation in a Model Organism
- In Vivo Functional Assay (Drosophila Example):
- System: Use a Drosophila model with tissue-specific Gal4/UAS system.
- Experimental Group: Express a human transgene (UAS-hEGFR) or a constitutively active form of the fly ortholog (UAS-Egfrλ) in a specific tissue (e.g., eye, using GMR-Gal4).
- Control Group: Cross driver line to a wild-type control (w1118).
- Readout: Image adult eyes using scanning electron microscopy (SEM) or brightfield microscopy. Quantify phenotypic severity (e.g., ommatidial disruption) using image analysis software (Fiji/ImageJ).
- Genetic Interaction: Cross the overexpression line with mutants in known pathway components (e.g., Ras85D, Mapk) to assess suppression/enhancement.
Diagram Title: Workflow for Cross-Species Hub Validation
Pathway Integration Diagram
A conserved signaling hub (e.g., EGFR/Ras/MAPK) links human disease data to model organism experimentation.
Diagram Title: Conserved EGFR/Ras/MAPK Hub Across Species
Table 3: Essential Reagents for Cross-Species Hub Analysis
| Reagent / Resource | Function in Hub Research | Example Source / Identifier |
|---|---|---|
| TCGAbiolinks R/Bioconductor Package | Facilitates programmatic download, integration, and analysis of TCGA multi-omics data. | Bioconductor Package |
| GEOquery R/Bioconductor Package | Retrieves and parses GEO data into R data structures for downstream analysis. | Bioconductor Package |
| SRA Toolkit | Command-line tools for downloading and converting SRA data to FASTQ for re-analysis. | NCBI GitHub |
| Alliance of Genome Resources API | Unified API to query orthology, gene function, and phenotypes across multiple MODs. | alliancegenome.org |
| Gal4/UAS System Lines (Drosophila) | Enables tissue-specific overexpression or RNAi of hub gene orthologs. | Bloomington Drosophila Stock Center (BDSC) |
| CRISPR/Cas9 Edited Mouse Lines | Knockout or knock-in models of hub genes for in vivo mammalian functional studies. | Knockout Mouse Project (KOMP) |
| Ortholog-Specific Antibodies | Validation of hub protein expression and localization in human and model organism tissues. | Commercial vendors (e.g., Abcam, DSHB) |
| Pathway Analysis Software (e.g., GSEA, Cytoscape) | Places candidate hub genes within biological pathways and interaction networks. | Broad Institute, Cytoscape.org |
The strategic integration of disease-specific data from TCGA, GEO, and SRA with the deep biological knowledge contained within Model Organism Databases creates a powerful engine for discovering and validating critical disease hubs. This multi-omics, cross-species approach, underpinned by the experimental protocols and resources outlined here, is essential for transforming genomic observations into mechanistically understood, therapeutically actionable targets.
In the context of multi-omics data repositories and resources research, integrating data from disparate molecular levels is paramount. Proteomics and metabolomics repositories serve as the foundational pillars for storing, sharing, and reanalyzing mass-spectrometry (MS) based data. These resources are critical for researchers and drug development professionals aiming to validate findings, perform meta-analyses, and build comprehensive systems biology models. This whitepaper provides an in-depth technical guide to four cornerstone repositories: PRIDE and PeptideAtlas for proteomics, and Metabolomics Workbench and MetaboLights for metabolomics.
The following table summarizes the core quantitative metrics and focal points of each repository, based on current data.
Table 1: Core Repository Specifications and Metrics
| Repository | Primary Focus | Data Types | Submission Format | Key Metrics (as of latest data) | Governing Body/Funding |
|---|---|---|---|---|---|
| PRIDE | Proteomics (MS) | Raw, processed, identification, quantification | mzML, mzIdentML, mzTab | >20,000 public datasets; >2.5 billion spectra | EMBL-EBI, ProteomeXchange Consortium |
| PeptideAtlas | Proteomics (MS) Spectral Library | Processed identifications, spectral libraries | mzIdentML, pepXML, mzTab | Builds for >30 organisms; billions of PSMs | Institute for Systems Biology (ISB) |
| Metabolomics Workbench | Metabolomics (MS & NMR) | Raw, processed, curated results | Study-specific templates, mzML, nmrML | >800 public studies; >500,000 chemical analyses | NIH Common Fund (USA) |
| MetaboLights | Metabolomics (MS & NMR) | Raw, processed, metadata | ISA-Tab, mzML, nmrML | >8,000 studies; >1.2 million metabolite assays | EMBL-EBI |
Detailed Technical Specifications and Access Protocols
PRIDE (Proteomics Identifications Database)
Mission: A centralized, public repository for MS-based proteomics data, supporting identification and quantification data.
- Access Protocol: Data is submitted via the ProteomeXchange (PX) consortium. The typical workflow involves:
- Preparation: Convert raw instrument files to open formats (e.g., .raw to .mzML using MSConvert from ProteoWizard).
- Metadata: Annotate the dataset using the PX submission tool with mandatory fields (sample details, protocol, instrument).
- Submission: Upload files via FTP or Aspera to the PRIDE server. A unique PX identifier (e.g., PXDxxxxxx) is issued.
- API Access: The PRIDE RESTful API (
https://www.ebi.ac.uk/pride/ws/archive/v2/) allows programmatic access to datasets, protein identifications, and spectral data.
PeptideAtlas
Mission: Provides a multi-organism, compendium of observed peptides from tandem MS experiments to support assay development and validation.
- Build Process Protocol: The creation of a PeptideAtlas build is a key computational experiment:
- Data Ingestion: Collect raw MS/MS data from public repositories (PRIDE, MassIVE).
- Uniform Reanalysis: Process all data through a consistent computational pipeline (e.g., the Trans-Proteomic Pipeline - TPP).
- Database Search: Search spectra against a target-decoy sequence database using search engines (e.g., Comet, X!Tandem).
- Statistical Validation: Apply statistical models (PeptideProphet, iProphet) to assign probabilities to peptide-spectrum matches (PSMs).
- Assembly: Filter high-confidence PSMs (e.g., ≥ 0.9 probability) and map to reference genomes to create a consolidated observability map.
Metabolomics Workbench
Mission: A US-based resource for metabolomics data, protocols, and analysis tools.
- Submission Protocol: The Metabolomics Workbench provides a structured submission system.
- Study Registration: Create a study with descriptive metadata (PI, publication, organism).
- Experimental Design: Define factors, groups, and sample relationships.
- Data Upload: Upload raw data files (instrument-specific or open mzML/nmrML) and processed data tables via a web interface.
- Chemical Annotation: Annotate metabolites using provided standards (HMDB, PubChem IDs) and describe identification confidence levels (Levels 1-4 as per COSMOS standards).
MetaboLights
Mission: A cross-species, cross-technique repository for metabolomics experiments.
- Submission and Curation Protocol: MetaboLights emphasizes rich metadata using the ISA (Investigation, Study, Assay) framework.
- ISA-Tab Creation: Use the ISAcreator tool to structure metadata into three linked files:
i_investigation.txt,s_study.txt,a_assay.txt. This captures the full experimental context from sample source to data generation. - Data Upload: Submit ISA-Tab files alongside raw and processed data files.
- Curation: Automated and manual curation checks for compliance and metadata completeness before public release (MTBLS identifier assigned).
- ISA-Tab Creation: Use the ISAcreator tool to structure metadata into three linked files:
Workflow and Relationship Visualizations
(Diagram 1: Proteomics Data Flow from Experiment to Public Resources)
(Diagram 2: Metabolomics Data Submission and Curation Pathways)
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Tools and Reagents for Repository-Centric Multi-Omics Research
| Item/Category | Function/Description | Example/Provider |
|---|---|---|
| Open Format Converters | Converts proprietary MS instrument data to open, community-standard formats for repository submission. | ProteoWizard MSConvert, nmrML converters |
| Metadata Annotation Tools | Software to create structured, standardized metadata required for high-quality repository submissions. | ISAcreator (MetaboLights), PX submission tool (PRIDE) |
| Spectral Search Engines | Core software for identifying peptides/metabolites from MS/MS spectra against sequence or chemical databases. | Comet, MaxQuant (Proteomics); MS-DIAL, Sirius (Metabolomics) |
| Statistical Validation Pipelines | Tools to assess confidence in identifications, filter false discoveries, and enable reproducible reanalysis. | Trans-Proteomic Pipeline (TPP), MzMine 3 (with Feature-Based Molecular Networking) |
| Reference Spectral Libraries | Curated collections of reference MS/MS spectra for peptide or metabolite identification. | NIST Tandem Mass Spectral Libraries, GNPS Public Spectra Libraries |
| Compound Databases | Structured chemical information for metabolite annotation and biological interpretation. | Human Metabolome Database (HMDB), PubChem, ChEBI |
| Programmatic Access Clients | Scripting packages to automate data retrieval, querying, and integration from repository APIs. | pyPRIDE, MetaboLightsR, jsonlite (for REST APIs) |
The integrated use of PRIDE, PeptideAtlas, Metabolomics Workbench, and MetaboLights is fundamental to advancing multi-omics research. They provide not just storage, but standardized frameworks, curated references, and programmatic access that transform disparate experimental data into reusable, collective knowledge. For drug development professionals, these repositories offer critical resources for biomarker validation, toxicology screening, and mechanistic elucidation. The future of systems biology relies on the continued evolution, interoperability, and adoption of these essential resources, guided by the FAIR principles (Findable, Accessible, Interoperable, Reusable).
Within the broader research thesis on Multi-omics data repositories, specialized portals that integrate genetic, proteomic, chemical, and cellular phenotypic data are critical for transforming systems biology insights into therapeutic hypotheses. LINCS, DepMap, and Pharos exemplify this evolution, providing curated, high-dimensional datasets and analytical tools that connect molecular perturbations to disease-relevant phenotypes. They serve as essential hubs for generating and validating hypotheses in target identification, lead optimization, and drug repurposing, embodying the translational power of integrated multi-omics resources.
The following table summarizes the core quantitative and functional attributes of each portal.
| Feature | LINCS (Library of Integrated Network-Based Cellular Signatures) | DepMap (Cancer Dependency Map) | Pharos (NIH Common Fund IDG Initiative) |
|---|---|---|---|
| Primary Focus | Cellular response signatures to chemical/genetic perturbations. | Genetic dependencies (CRISPR screens) & biomarkers in cancer models. | Annotation and prioritization of understudied drug targets. |
| Core Data Type | L1000 transcriptomics, proteomics, cell imaging, kinase activity. | CRISPR knockout viability, RNAi, CNV, gene expression, methylation. | Knowledge graph integrating Target Development Level (TDL), literature, drugs, pathways. |
| Scale (as of 2024) | ~2M gene expression profiles; ~50k perturbagens; 100+ cell lines. | 1,800+ cancer cell lines; 18,000+ genes screened; 1,100+ molecular datasets. | ~20,000 human protein targets; ~1.5M bioactivities; 500,000+ publications mined. |
| Key Output | Connectivity maps, signature similarity, network models. | Dependency scores (Chronos), biomarkers, gene effect scores. | TDL classification, disease associations, ligandability, GO annotations. |
| Primary Application | Mechanism of action discovery, drug repurposing, pathway analysis. | Target identification, biomarker discovery, synthetic lethality. | Target prioritization, feasibility assessment, knowledge gap identification. |
Detailed Methodologies and Experimental Protocols
LINCS L1000 Transcriptomic Profiling Protocol
This high-throughput, low-cost method infers the expression of ~12,000 genes from a measured set of 978 "landmark" genes.
Protocol Steps:
- Cell Seeding & Perturbation: Seed cells in 384-well plates. Treat with small molecule compounds (at multiple doses) or introduce genetic perturbations (e.g., siRNA).
- Lysis & mRNA Capture: After incubation (typically 24-48h), lyse cells and isolate mRNA using bead-based capture.
- Ligation-Mediated Amplification:
- Reverse Transcription: Convert mRNA to cDNA with gene-specific primers.
- Ligation: Add a universal adapter via ligation.
- PCR Amplification: Amplify cDNA with fluorescently-labeled universal PCR primers.
- Detection & Quantification: Hybridize amplified material to Luminex beads. Measure fluorescence intensity for each landmark gene.
- Data Inference (CLUE Platform): Use a computational model (trained on full transcriptome data) to infer the expression of ~12,000 non-measured "imitating" genes from the landmark gene profile.
- Signature Generation & Connectivity: Generate differential expression signatures (perturbed vs. control). Query signatures against the LINCS database via the CLUE platform to find connections between perturbagens with similar or opposite signatures.
DepMap CRISPR-Cas9 Knockout Screening Protocol
This protocol identifies genes essential for cancer cell survival and proliferation (genetic dependencies).
Protocol Steps:
- Library Design: Use the Brunello or similar genome-wide sgRNA library (4-6 sgRNAs per gene, plus non-targeting controls).
- Virus Production: Lentivirally package the sgRNA library in HEK293T cells.
- Cell Infection & Selection: Infect a pool of cancer cells (e.g., A549) at low MOI to ensure single integration. Select with puromycin for 72+ hours. This is the initial timepoint (T0).
- Cell Passaging: Culture cells for ~18-21 population doublings, maintaining representation of >500 cells per sgRNA.
- Genomic DNA Extraction & Sequencing: Harvest cells at T0 and at the final timepoint (Tend). Extract gDNA, amplify integrated sgRNA sequences via PCR, and sequence using next-generation sequencing.
- Dependency Score Calculation (Chronos Algorithm): Count sgRNA reads. The Chronos algorithm models read count depletion, accounting for screen noise, copy-number effects, and variable sgRNA activity. It outputs a gene effect score (negative scores indicate essentiality; typically, a score < -1 suggests strong dependency).
Visualizations
Title: LINCS L1000 Experimental and Computational Workflow
Title: DepMap Data Integration for Target Discovery
The Scientist's Toolkit: Essential Research Reagents & Materials
| Reagent / Resource | Function in Protocol |
|---|---|
| L1000 Luminex Bead Kit | Enables multiplexed quantification of 978 landmark gene transcripts. |
| Brunello sgRNA Library | Genome-wide CRISPR knockout library (4 sgRNAs/gene) used in DepMap screens. |
| Chronos Algorithm (Software) | Computes gene dependency scores from CRISPR screen read counts, correcting for confounders. |
| CLUE Platform (clue.io) | Web interface for querying LINCS signatures and computing connectivity. |
| Pharos Knowledge Graph API | Programmatic access to integrated target annotations for custom analysis pipelines. |
| DepMap Public 23Q4+ Dataset | Pre-processed dependency matrices and multi-omics data for all characterized cell lines. |
| HT-29 or A549 Cell Lines | Commonly used cancer cell models in both LINCS (perturbation) and DepMap (dependency) studies. |
| Lentiviral Packaging Plasmids | psPAX2 and pMD2.G for producing lentivirus in CRISPR screening workflows. |
In the context of multi-omics data repositories and resources research, the integration and interpretation of complex biological datasets demand rigorous metadata standards. The ISA (Investigation, Study, Assay) framework, the Minimum Information for Biological and Biomedical Investigations (MIBBI), and the FAIR (Findable, Accessible, Interoperable, Reusable) principles collectively form the cornerstone of reproducible and integrative systems biology. This whitepaper details their technical implementation, methodologies for compliance, and their indispensable role in modern drug development and translational research.
Core Frameworks and Standards
1.1 ISA-Tab and ISA-Tools The ISA framework structures experimental metadata using a hierarchical, tab-delimited format (ISA-Tab). The open-source ISA software suite facilitates the creation, curation, and management of ISA-Tab files.
Key Components:
- Investigation: The overarching project context.
- Study: A unit of research with defined objectives.
- Assay: A specific analytical measurement.
Experimental Protocol for ISA Metadata Curation:
- Define Experimental Design: Outline all factors, variables, and experimental units.
- Install ISAcreator: Download and configure the Java-based ISAcreator tool.
- Populate ISA-Templates: For each assay type (e.g., LC-MS metabolomics, RNA-seq), use the guided interface to input:
- Source Name, Characteristics
- Protocol steps with parameters (e.g., "nucleic acid extraction", instrument model)
- Raw and derived data file names.
- Validate and Export: Use the internal validator to check for missing mandatory fields, then export as ISA-Tab.
- Conversion: Utilize the
isatab2jsonorisatab2uploadcommands to prepare submissions for repositories like MetaboLights or ArrayExpress.
1.2 MIBBI and Reporting Guidelines MIBBI serves as a portal to over 40 Minimum Information checklists (e.g., MIAME for microarray, MIAPE for proteomics). Adherence ensures the scientific community can critically evaluate and reproduce experimental results.
- Protocol for MIBBI-Compliant Reporting:
- Checklist Selection: Identify all relevant checklists for a multi-omics study (e.g., MINSEQE for sequencing, MSI for metabolomics).
- Metadata Audit: Cross-reference experimental records against each checklist's required data elements.
- Gap Analysis and Remediation: Document and address any missing information before public deposition.
1.3 The FAIR Guiding Principles FAIR principles provide a metrics-oriented framework for data stewardship, emphasizing machine-actionability.
Table 1: Impact of Metadata Standards on Data Reusability Metrics
| Metric | Pre-Standard Implementation (Baseline) | Post ISA/FAIR Implementation (Reported Improvement) | Source / Study Context |
|---|---|---|---|
| Data Findability (Repository Search Success Rate) | ~35% | ~85% | Analysis of curated vs. uncurated submissions in EBI repositories |
| Process Automation (Manual Curation Time per Dataset) | 8-12 hours | 1-2 hours | Internal benchmarking at a major pharma consortium |
| Multi-omics Integration Success Rate | ~25% | ~78% | Review of 50+ integrated studies in systems pharmacology |
Table 2: Core MIBBI Checkpoints for Multi-omics
| Omics Layer | Primary MIBBI Checklist | Critical Required Metadata Fields (Examples) |
|---|---|---|
| Genomics/Transcriptomics | MINSEQE | Read length, sequencing platform, alignment software name/version, processed data file format. |
| Proteomics | MIAPE | Instrument configuration, dissociation method, search engine parameters, false discovery rate threshold. |
| Metabolomics | MSI | Sample extraction method, chromatography type, mass analyzer, metabolite identification confidence. |
Implementing a FAIR Multi-omics Workflow
Detailed Experimental Protocol: From Bench to Repository
Pre-Experimental Planning:
- Register the study in a persistent registry (e.g.,
doi.org/10.21228/...) to obtain a unique, machine-readable identifier (F1). - Define a comprehensive metadata capture plan using relevant MIBBI checklists.
- Register the study in a persistent registry (e.g.,
Data & Metadata Generation:
- Execute omics assays per SOPs.
- Concurrently, populate an ISA-Tab structure via ISAcreator, linking each data file to detailed protocols and sample characteristics.
Curation & Validation:
- Run the
isatab2jsonconverter and validate the resulting JSON against the ISA-JSON schema. - Use FAIR evaluation tools (e.g., FAIRplus SAFE tool) to generate a compliance score.
- Run the
Deposition & Publication:
- Submit the ISA archive and raw data to a certified repository (e.g., PRIDE for proteomics, GEO for transcriptomics).
- Ensure the publication references both the data DOI and the metadata DOI.
Visualizing the Metadata Ecosystem
Diagram 1: The Metadata Management Lifecycle in Multi-omics Research
Diagram 2: ISA Framework Enabling Multi-omics Data Integration
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Tools and Resources for Metadata Management
| Item / Resource | Function / Role | Example (Vendor/Project) |
|---|---|---|
| ISAcreator Software | Desktop application for generating and managing ISA-Tab metadata. | ISA-Tools GitHub Repository |
| FAIR Evaluator | Web service to assess the FAIRness of a digital resource. | FAIRplus SAFE Tool |
| BioSamples Database | Repository to assign unique, persistent IDs to biological samples. | EMBL-EBI BioSamples |
| Protocols.io | Platform for detailing, sharing, and versioning experimental protocols with DOIs. | Protocols.io |
| Ontology Lookup Service (OLS) | Service to find and use standardized ontological terms for metadata. | EMBL-EBI OLS |
| MIBBI Portal | Registry to identify and consult relevant minimum information checklists. | FAIRsharing.org (hosts MIBBI legacy) |
| ISA-JSON Configuration | Schema files defining the structure for machine-readable ISA metadata. | ISA Model (JSON Schema) on GitHub |
From Data to Discovery: Practical Strategies for Accessing, Integrating, and Analyzing Multi-omics Resources
The integration of diverse omics data—genomics, transcriptomics, proteomics, and metabolomics—is foundational to modern systems biology and precision medicine. A central challenge in multi-omics research is the programmatic aggregation, normalization, and analysis of data dispersed across specialized, heterogeneous repositories. This whitepaper provides a technical guide for researchers to leverage application programming interfaces (APIs) and specialized R packages to overcome these barriers, enabling reproducible, large-scale data retrieval and integration essential for robust multi-omics thesis research.
Core APIs for Bioinformatics Data Retrieval
NCBI E-utilities (Entrez Programming Utilities)
NCBI's E-utilities provide a stable interface to query and retrieve data from over 40 databases, including PubMed, Gene, SRA, and dbSNP. They are essential for fetching genomic and literature data.
Key Operations:
- EInfo: Obtain database statistics.
- ESearch: Perform text searches, returning primary ID lists.
- EFetch: Retrieve full records in various formats (XML, FASTA, etc.).
Current Quantitative Summary (Live Search Data):
| Database | Estimated Records (Approx.) | Key Data Type | Update Frequency |
|---|---|---|---|
| PubMed | 36+ million citations | Biomedical literature | Daily |
| SRA | 45+ million experiments | Raw sequencing data | Continuous |
| Gene | 70+ million entries | Gene-centric data | Weekly |
| Protein | 300+ million sequences | Protein sequences | Daily |
| dbSNP | 2+ billion submitted SNPs | Genetic variation | Continuous |
Protocol 1: Programmatic Gene Data Retrieval via E-utilities
- Construct the ESearch URL: Find gene IDs for a query (e.g., "TP53 human"). Base URL:
https://eutils.ncbi.nlm.nih.gov/entrez/eutils/esearch.fcgi - Parameters:
db=gene&term=TP53[gene]+AND+human[orgn]&retmode=json - Parse Response: Extract the list of Gene IDs (e.g.,
7157) from the JSON result. - Construct the EFetch URL: Retrieve detailed records. Base URL:
https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi - Parameters:
db=gene&id=7157&retmode=xml - Parse XML Output: Extract fields like genomic location, aliases, and summaries using an XML parser.
EMBL-EBI BioServices and Web APIs
The EMBL-EBI offers RESTful APIs for its vast resources, often providing more direct access to bio-specific data formats compared to E-utilities.
Key Resources:
- Ensembl REST API: Access genomic features, sequences, variants, and comparative genomics.
- UniProt REST API: Retrieve protein sequences, functional annotations, and variant data.
- OMEGA API (for Metabolomics): Access chemical structures and related biological data.
Protocol 2: Fetching Protein Information via UniProt API
- Define Accession: Identify target protein (e.g.,
P04637for human TP53). - Construct Request URL:
https://www.ebi.ac.uk/proteins/api/proteins/P04637 - Set Headers: Include
Accept: application/jsonin the HTTP request header. - Send GET Request: Use tools like
curl,requests(Python), orhttr(R). - Parse JSON Response: Extract relevant fields from the nested JSON structure (e.g.,
gene.name,protein.recommendedName.fullName,features).
R/Bioconductor for Integrated Multi-omics Analysis
Bioconductor provides over 2,000 packages for the analysis and comprehension of high-throughput genomic and multi-omics data, emphasizing reproducibility and statistical rigor.
Core Packages for Data Retrieval and Integration
| Package Name | Primary Function in Multi-omics Workflow | Key Data Source Integration |
|---|---|---|
rentrez |
Wrapper for NCBI E-utilities; searches and downloads records. | PubMed, Gene, SRA, dbSNP |
biomaRt |
Interfaces with Ensembl BioMart; maps gene IDs, gets sequences. | Ensembl genomes |
AnnotationHub| Manages and retrieves large collection of genome-wide annotations. |
UCSC, Ensembl, ENCODE | |
GEOquery |
Downloads and parses Gene Expression Omnibus (GEO) data. | NCBI GEO |
MultiAssayExperiment| Integrates multiple experimental assays on shared specimen collections. |
User-provided multi-omics data |
Protocol 3: Multi-omics ID Mapping and Annotation with biomaRt
- Load Library and Connect to Mart:
ensembl <- useMart("ensembl") - Select Dataset:
ensembl <- useDataset("hsapiens_gene_ensembl", mart = ensembl) - Define Attributes/Filters: Specify desired output (e.g.,
c('entrezgene_id', 'hgnc_symbol', 'ensembl_transcript_id')) and input filter (e.g.,'entrezgene_id'). - Run Query:
getBM(attributes = attributes_list, filters = 'entrezgene_id', values = my_gene_list, mart = ensembl) - Merge Results: Integrate the annotation table with expression or variant data using common identifiers.
Protocol 4: Creating a Multi-omics Data Container with MultiAssayExperiment
- Prepare Assays as Lists: Organize your separate omics data matrices (e.g., RNA-seq, methylation) into a named
list. Ensure column names (samples) are consistent. - Prepare Sample Metadata: Create a
DataFramewhere rows correspond to samples and columns describe sample phenotypes. - Prepare Feature Metadata: Provide a
DataFramefor each assay describing the features (e.g., gene annotations). - Construct Object:
myMAE <- MultiAssayExperiment(experiments = assay_list, colData = sample_metadata, maps = feature_metadata_list) - Subset and Analyze: Use
myMAE[, , "assay_name"]to subset and apply assay-specific statistical methods.
Visualization of Workflows and Relationships
Diagram 1: Multi-omics Data Retrieval and Integration Workflow
Diagram 2: Logical Relationship of Key R/Bioconductor Packages
The Scientist's Toolkit: Essential Research Reagent Solutions
| Item / Resource | Function in Programmatic Multi-omics Research |
|---|---|
| RStudio IDE | Integrated development environment for R, facilitating script writing, visualization, and package management. |
| BiocManager | The primary R package used to install and manage Bioconductor packages and their dependencies. |
httr / curl R Packages |
Provide powerful tools for constructing, sending, and handling HTTP requests to web APIs (e.g., E-utilities, EMBL-EBI). |
jsonlite / xml2 R Packages |
Essential parsers for converting API responses (JSON/XML) into structured R data objects (lists, data.frames). |
| Jupyter / R Notebooks | Environments for creating literate programming documents that combine executable code, results, and narrative text, ensuring full reproducibility. |
| Git & GitHub | Version control system and platform for tracking code changes, collaborating, and sharing analysis pipelines. |
| Docker / Bioconductor Docker Images | Containerization technology that packages an analysis environment (OS, R, packages), guaranteeing identical and reproducible runtime conditions. |
The exponential growth of multi-omics data—from genomics, transcriptomics, proteomics, and metabolomics—presents both an unprecedented opportunity and a significant computational challenge in biomedical research and drug development. Traditional on-premises infrastructure often lacks the scalability, elasticity, and collaborative features required to manage petabytes of data and perform complex, integrative analyses. This whitepaper, framed within a broader thesis on multi-omics data repositories and resources, provides an in-depth technical guide to four leading cloud-based platforms: Terra, BioData Catalyst, Seven Bridges, and Google Cloud. We examine their architectures, capabilities, and applications for enabling scalable, reproducible, and collaborative analysis.
Platform Architecture and Feature Comparison
The following table summarizes the core architectural components, primary funding agencies, and key distinguishing features of each platform.
Table 1: Core Platform Comparison
| Platform | Lead Organization / Funders | Core Cloud Backend | Primary Data Repositories | Key Distinguishing Feature |
|---|---|---|---|---|
| Terra | Broad Institute (NIH, Google) | Google Cloud, Azure | AnVIL, Gen3, BioData Catalyst | "Bring Your Own Tools" flexibility; Jupyter/R Studio integration |
| BioData Catalyst | NHLBI (NIH) | Google Cloud, AWS | TOPMed, dbGaP, GEO | Ecosystem focused on NHLBI data; federated authentication |
| Seven Bridges | Seven Bridges Genomics | AWS, Google Cloud, Azure | CRL, TCGA, ICA | Commercial platform with strong focus on pipeline portability (CWL) |
| Google Cloud | Google Cloud | Public Datasets, Biogenetics | Raw IaaS/PaaS; maximal configurability and ML/AI integration |
Quantitative Performance and Cost Metrics
Performance benchmarks vary based on workload, but the following table provides a generalized comparison based on published use cases.
Table 2: Performance and Cost Indicators (Approximate)
| Platform | Typical WGS Alignment Time (100x coverage) | Approximate Cost per WGS Analysis* | Built-in Workflow Languages | Native Integration with AI/ML Tools |
|---|---|---|---|---|
| Terra | 4-6 hours | $25-$40 | WDL, CWL, Nextflow | Yes (Google Vertex AI, Galaxy) |
| BioData Catalyst | 5-7 hours | $30-$45 | WDL, CWL, Jupyter Notebooks | Limited |
| Seven Bridges | 4-5 hours | $35-$50 | CWL, WDL | Yes (Built-in ML tools) |
| Google Cloud | 3-5 hours | $20-$60 (highly configurable) | Any (DIY) | Yes (Vertex AI, TensorFlow, BigQuery ML) |
*Cost estimates include compute, storage I/O, and data egress for a standard GATK Best Practices pipeline, using comparable VM instances. Actual costs are highly workload-dependent.
Experimental Protocol: A Scalable Multi-omics Integration Analysis
This protocol details a representative cloud-based analysis integrating genomic and transcriptomic data to identify driver mutations and their functional transcriptional consequences.
Title: Cloud-Native Somatic Variant Calling and Differential Expression Analysis
Objective: To identify somatic variants from paired tumor-normal whole genome sequencing (WGS) and correlate findings with tumor RNA-seq differential expression data.
Platform-Setup (Generalized):
- Data Acquisition & Cohorting: Use the platform's data portal (e.g., Terra Data Library, BioData Catalyst PIC-SURE API) to select and create a cohort of paired WGS (normal vs. tumor) and RNA-seq BAM files from a repository like The Cancer Genome Atlas (TCGA).
- Workspace Configuration: Create a new analysis workspace. Configure cloud compute profiles (e.g., Google Cloud project, pre-emptible VM settings) and attach secure, credentialed access to the selected data.
- Data Processing - Genomics:
- Tool: GATK4 Mutect2 (via Broad's optimized WDL pipeline).
- Input: CRAM/BAM files (aligned reads).
- Process: Launch the "Somatic-SNVs-Indels-GATK4" WDL workflow from the platform's Methods Repository. Specify reference genomes (hg38), genomic intervals, and required databases (gnomAD, dbSNP).
- Output: VCF file of somatic variants, annotated with Funcotator.
- Data Processing - Transcriptomics:
- Tool: STAR for alignment, DESeq2 (via R in a Jupyter Notebook) for differential expression.
- Input: RNA-seq FASTQ files.
- Process: Launch the "RNA-seq Alignment and Expression Quantification" CWL workflow. Use the resulting count matrix as input for a cloud-hosted RStudio session running DESeq2 to compare tumor vs. normal expression.
- Output: Normalized count matrix and list of differentially expressed genes (DEGs).
- Integrative Analysis:
- Tool: Custom Python/R script in a Jupyter Notebook.
- Process: Load the somatic VCF and DEG list. Perform statistical enrichment (e.g., using Fisher's exact test) to check if genes harboring somatic variants are overrepresented among DEGs. Visualize results (e.g., Manhattan plots, heatmaps).
- Reproducibility & Sharing: Package the entire workspace—including data references, workflow configurations, parameters, and interactive notebooks—and share it with collaborators via the platform's access controls.
Diagram: Multi-omics Cloud Analysis Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Analytical "Reagents" for Cloud-Based Multi-omics Analysis
| Item / Solution | Function in Analysis | Example (Platform Specific) |
|---|---|---|
| Workflow Definition Language (WDL/CWL) | Defines the computational pipeline (tools, steps, resources) for portability and reproducibility. | Broad's GATK WDLs (Terra), CWL tool definitions (Seven Bridges) |
| Docker Container Images | Provides a standardized, isolated software environment for each analytical tool. | biowdl/gatk:latest, quay.io/biocontainers/star:2.7.10a |
| Cloud-Optimized File Formats | Enables efficient, partial data access (query) without downloading entire files. | CRAM (for reads), Google Genomics VCF, TileDB |
| Interactive Analysis Notebook | Allows for exploratory data analysis, visualization, and custom scripting in a shared environment. | JupyterLab (Terra, BioData Catalyst), RStudio (Seven Bridges) |
| Data Access and Query Layer | Provides secure, programmatic access to controlled and public data without manual transfer. | Gen3 Indexd & Fence (BioData Catalyst), DRAGEN API (Google Cloud) |
| Benchmarking & Cost Estimator | Predicts runtime and cost for a workflow given specific parameters, aiding in budget planning. | Seven Bridges CODA, Google Cloud Pricing Calculator |
The choice of a cloud platform for multi-omics analysis hinges on specific research needs. Terra excels in open, collaborative science with extreme flexibility in tool choice. BioData Catalyst is optimized for researchers deeply embedded in NHLBI-funded studies and data. Seven Bridges provides a highly supported, commercial-grade environment with strong compliance frameworks. Google Cloud offers the deepest level of control and integration with cutting-edge AI services for teams with strong engineering support.
The future of multi-omics research is inextricably linked to cloud-native ecosystems that unify data, computing, and collaboration. Success requires investing not only in infrastructure but also in skills for workflow languages, data management, and cost optimization. These platforms democratize access to scalable computational power, accelerating the translation of massive biological datasets into actionable insights for drug discovery and precision medicine.
The integration of genomics, transcriptomics, proteomics, and metabolomics data—multi-omics—is fundamental for advancing systems biology and precision medicine. A core challenge in this domain is the reproducible and scalable processing of heterogeneous, high-volume data. This technical guide examines three pivotal workflow management systems—Galaxy, Nextflow, and Snakemake—as engines for building robust, reproducible analysis pipelines essential for multi-omics data repositories and resources research.
Table 1: Quantitative Comparison of Workflow Systems in Multi-omics Context
| Feature | Galaxy | Nextflow | Snakemake |
|---|---|---|---|
| Primary Language | Graphical UI / XML | DSL (Groovy-based) | Python-based DSL |
| Execution Environment | Conda, Docker, Singularity | Docker, Singularity, Conda, Podman | Conda, Docker, Singularity, Apptainer |
| Portability | High (via Platform) | Very High (Self-contained) | Very High (Self-contained) |
| Scaling Architecture | Clusters, Cloud (via Plugins) | Built-in for HPC, Kubernetes, Cloud | HPC, Cloud (via Profiles) |
| Key Strength | Accessibility, Tool Discovery | Scalability, Stream-oriented | Python Integration, Readability |
| 2024 Community Tools (BioConda) | ~9,800 | ~3,200 (pipelines) | ~2,800 (rules) |
| Typical Use Case | Accessible, Shared Platform | Large-scale, Distributed Pipelines | Complex, Python-centric Analyses |
Detailed Methodologies for Pipeline Implementation
Protocol: Building a Cross-Platform RNA-Seq Pipeline
This protocol outlines steps to create a reproducible RNA-seq analysis pipeline, adaptable across all three systems.
A. Initial Setup and Dependency Management
- Environment Isolation: For all systems, begin by defining software dependencies. Use BioConda to create an
environment.yamlfile listing packages (e.g.,fastp=0.23.4,salmon=1.10.1,multiqc=1.19). - Containerization: For maximal reproducibility, build or pull Docker/Singularity images containing all dependencies. Nextflow and Snakemake natively support pulling containers per process/rule. Galaxy tools integrate containers via the Tool Definition.
B. Workflow Definition
- In Galaxy: Use the graphical editor to chain tools:
FASTQ Input→Fastp (trimming)→Salmon (quantification)→MultiQC (reporting). Export the workflow as a.gafile or represent it informat 2Galaxy Tool Definition Language. - In Nextflow: Create a
main.nffile. Define processes for each step (trim, quantify, aggregate) and channel-based inputs/outputs. - In Snakemake: Create a
Snakefile. Define rules with input/output wildcards and conda/container directives.
C. Execution and Reproducibility
- Galaxy: Execute on a public server (usegalaxy.org), a private instance, or via the command line using
planemo run. - Nextflow: Run with
nextflow run main.nf -profile docker,cluster. The-profilesystem manages configuration for different executors. - Snakemake: Execute with
snakemake --use-conda --use-singularity --cores 8. The--profileflag can apply pre-defined cluster configurations.
D. Provenance Capture
All three systems automatically generate provenance information: Galaxy in its database and via researchobject bundles, Nextflow in a trace report and execution timeline, Snakemake in a run report and conda environment logs.
Visualizing Workflow Integration Patterns
Title: Integration Pattern for Multi-omics Pipeline Execution
Title: Researcher Decision Path for Reproducible Analysis
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 2: Key Research Reagent Solutions for Multi-omics Pipeline Development
| Item / Resource | Function in Workflow Integration | Example / Source |
|---|---|---|
| BioConda | Provides versioned, interoperable bioinformatics packages for all three workflow systems. | https://bioconda.github.io/ |
| BioContainers | Supplies ready-to-use Docker/Singularity containers for BioConda packages, ensuring environment consistency. | https://biocontainers.pro/ |
| CWL / WDL Exporters | Enables conversion of workflows to Common Workflow Language (CWL) or Workflow Description Language (WDL) for cross-platform execution. | Galaxy's gxformat2, snakemake --export-cwl, Nextflow's cwl-export plugin. |
| Workflow Hub | A registry for sharing, publishing, and executing FAIR (Findable, Accessible, Interoperable, Reusable) computational workflows. | https://workflowhub.eu/ |
| MultiQC | Aggregates results from numerous bioinformatics tools into a single interactive report, a common final step in omics pipelines. | https://multiqc.info/ |
| Research Object Bundler | Packages workflow, code, data, and provenance into a reproducible, citable archive. | ro-crate tools integrated in Galaxy, nextflow logs. |
| Institutional HPC/Cloud Scheduler | Provides the execution backbone for scalable processing (SLURM, AWS Batch, Google Life Sciences). | Required for leveraging the parallel power of Nextflow/Snakemake. |
Within the broader context of multi-omics data repositories and resources research, the integration of disparate molecular data layers—genomics, transcriptomics, proteomics, metabolomics—is paramount for holistic biological understanding and drug discovery. Data fusion tools transform heterogeneous repositories into coherent, actionable insights. This technical guide provides an in-depth analysis of leading integration frameworks, focusing on MOFA and mixOmics, their methodologies, and applications in biomedical research.
Core Integration Frameworks: A Comparative Analysis
MOFA/MOFA+: Multi-Omics Factor Analysis
MOFA is a Bayesian framework that uses Factor Analysis to decompose multi-omics data into a set of latent factors representing the shared sources of variation across data types.
Key Algorithmic Steps:
- Model Specification: For each data modality m, the model assumes: X^m = Z W^{m^T} + ε^m, where X is the data matrix, Z is the latent factor matrix, W^m are modality-specific weights, and ε^m is noise.
- Variational Inference: A scalable inference algorithm approximates the posterior distributions of all model parameters (Z, W).
- Factor Interpretation: Latent factors are correlated with sample metadata (e.g., clinical outcome) and annotated via inspection of highly weighted features.
Experimental Protocol for Applying MOFA+ (R/Python):
- Input Data Preparation: Normalize and scale each omics dataset (e.g., RNA-seq counts, methylation beta-values). Ensure matched samples.
- Model Training: Run
MOFAobject <- create_mofa(data)andMOFAobject <- run_mofa(MOFAobject)with options for factor number (automatic or user-defined), sparsity (ARD priors), and convergence tolerance. - Downstream Analysis: Use functions like
plot_variance_explained(MOFAobject)andcorrelate_factors_with_covariates(MOFAobject, metadata). - Biological Interpretation: Extract top features per factor per view (
get_weights(MOFAobject)) for pathway enrichment analysis (e.g., via g:Profiler).
mixOmics: Multivariate Exploratory Data Analysis
mixOmics provides a suite of multivariate methods (e.g., PLS, CCA, DIABLO) for dimension reduction and integration, emphasizing discriminative analysis for supervised problems like classification.
Key Method: DIABLO (Data Integration Analysis for Biomarker discovery using Latent variable approaches)
- Objective: Identify highly correlated multi-omics features that discriminate between predefined sample groups.
- Algorithm: A generalization of multi-block PLS-DA. It maximizes covariance between latent components from each data block while also maximizing discrimination between groups.
- Tuning: Critical step is tuning the number of components and the design matrix, which controls the inter-omics block correlation strength.
Experimental Protocol for DIABLO:
- Data Setup: Organize omics matrices into a list. Define Y as a factorial outcome vector (e.g., disease state).
- Parameter Tuning: Use
tune.block.splsda()to perform repeated cross-validation and select the number of features to keep per dataset and per component, and the design value. - Model Building: Run final model with
block.splsda(X, Y, ncomp, keepX, design). - Evaluation & Output: Assess performance with
perf()(cross-validated error rates). Plot integrated sample clusters (plotIndiv) and select driving features (plotLoadings,selectVar).
Additional Noteworthy Frameworks
- Integrative NMF (iNMF): Uses Non-negative Matrix Factorization to identify shared and dataset-specific factors.
- Similarity Network Fusion (SNF): Constructs sample-similarity networks for each data type and fuses them into a single network for clustering.
- JIVE (Joint and Individual Variation Explained): Decomposes data into joint variation across all types and individual variation specific to each type.
Quantitative Framework Comparison
Table 1: Core Characteristics of Multi-omics Integration Tools
| Feature | MOFA/MOFA+ | mixOmics (DIABLO) | iNMF | SNF |
|---|---|---|---|---|
| Core Methodology | Bayesian Factor Analysis | Multi-block PLS-DA (supervised) | Non-negative Matrix Factorization | Network Fusion & Spectral Clustering |
| Primary Goal | Uncover hidden sources of variation | Supervised classification & biomarker ID | Identify shared & specific patterns | Sample clustering via network fusion |
| Data Input | Any numeric, matched samples | Any numeric, matched samples | Any non-negative, matched samples | Any numeric, matched samples |
| Handling of Missing Data | Yes (probabilistically) | No (requires imputation) | Limited | Yes (within-network calculation) |
| Key Output | Latent factors, variance explained | Latent components, selected features, classification performance | Feature modules (shared/specific) | Fused sample network, clusters |
| Typical Use Case | Exploratory analysis of population heterogeneity | Predicting clinical outcome from multi-omics | Decomposing co-regulation patterns | Cancer subtype discovery |
Table 2: Statistical & Software Attributes
| Attribute | MOFA/MOFA+ | mixOmics (DIABLO) |
|---|---|---|
| Inference Method | Variational Bayesian | Partial Least Squares optimization |
| Sparsity Control | Automatic Relevance Determination (ARD) | L1 penalization (keepX parameter) |
| Programming Language | R, Python | R |
| Critical Parameter to Tune | Number of factors (can be auto-inferred) | Number of components, keepX, design matrix |
| Primary Visualization | Variance explained plots, factor scatterplots | Sample plot, loadings plot, circos plot |
Visualizing Workflows and Relationships
Diagram 1: MOFA+ Analysis Pipeline
Diagram 2: DIABLO Supervised Analysis Path
Diagram 3: Tool Selection by Analysis Goal
Table 3: Key Reagents & Computational Resources for Multi-omics Integration
| Item | Category | Function in Multi-omics Fusion |
|---|---|---|
| Reference Multi-omics Datasets (e.g., TCGA, GTEx, Depression Cohort) | Data Resource | Provide matched, clinically annotated omics data for method benchmarking and discovery. |
| High-Throughput Sequencing Kits (RNA-seq, WGBS, ATAC-seq) | Wet-lab Reagent | Generate foundational genomics/transcriptomics data layers for integration. |
| Mass Spectrometry Reagents (TMT/Isobaric Tags, LC Columns) | Wet-lab Reagent | Enable quantitative proteomics and metabolomics data generation. |
R/Bioconductor MOFA2 Package |
Software Tool | Implements the MOFA+ model for flexible, unsupervised integration in R. |
R mixOmics Package |
Software Tool | Provides DIABLO and other multivariate methods for supervised/unsupervised integration. |
Python mofapy2 Package |
Software Tool | Python implementation of the MOFA model for integration into Python workflows. |
| Pathway Enrichment Tools (g:Profiler, clusterProfiler, MetaboAnalyst) | Software Resource | Biologically interpret feature sets identified by integration tools. |
| High-Performance Computing (HPC) Cluster or Cloud (AWS, GCP) | Computational Resource | Enables analysis of large-scale multi-omics data, which is computationally intensive. |
The choice of fusion tool—whether MOFA+ for unsupervised discovery of latent factors, mixOmics/DIABLO for supervised biomarker identification, or SNF for robust clustering—is dictated by the specific biological question and data structure. As multi-omics repositories grow in scale and complexity, these frameworks are essential for translating molecular data into mechanistic insights and therapeutic targets, forming a critical component of modern computational biology and precision medicine research.
Thesis Context: This technical guide is framed within a broader thesis on Multi-omics data repositories and resources research, focusing on integrative analysis to derive actionable biological insights.
The integration of transcriptomic and proteomic data is a cornerstone of multi-omics research, offering a more comprehensive view of biological systems than any single layer can provide. Public repositories house vast amounts of such data, but their disparate nature poses significant analytical challenges. This guide details a systematic approach for correlating these datasets to identify robust candidate biomarkers for diseases like cancer or neurodegenerative disorders.
Key Public Data Repositories
The following table summarizes the primary repositories used in such integrative studies.
Table 1: Primary Public Repositories for Transcriptomic and Proteomic Data
| Repository Name | Data Type | Primary Focus | Typical Data Format | Access Method |
|---|---|---|---|---|
| Gene Expression Omnibus (GEO) | Transcriptomic (RNA-seq, microarray) | Curated gene expression profiles | SOFT, MINiML, raw FASTQ/BAM | Web interface, GEOquery (R) |
| Sequence Read Archive (SRA) | Transcriptomic (Raw sequencing reads) | Raw sequencing data for reprocessing | FASTQ, BAM | SRA Toolkit, web browser |
| ProteomeXchange Consortium | Proteomic (Mass spectrometry) | Coordinated submission of proteomics datasets | mzML, mzIdentML, raw vendor files | Via member repositories (PRIDE, MassIVE) |
| PRIDE Archive | Proteomic (Mass spectrometry) | Functional proteomics data repository | mzML, mzIdentML | Web API, rpx (R) |
| CPTAC Data Portal | Proteomic, Transcriptomic (Cancer-focused) | Pre-processed, harmonized cancer multi-omics data | TSV, BED, processed matrices | Web portal, Gen3 SDK |
| dbGaP | Phenotype & Genotype | Clinical data linked to molecular data (controlled access) | Various, subject to authorization | Controlled access request |
Core Experimental and Analytical Protocol
Protocol: Data Acquisition and Harmonization
- Define Cohort: Specify disease of interest, tissue type, sample size requirements, and clinical parameters (e.g., tumor vs. normal, disease stage).
- Repository Query: Use repository-specific search terms (e.g., "glioblastoma," "Homo sapiens," "tumor tissue," "RNA-seq," "LC-MS/MS"). Leverage metadata filters for instrument platform, sample preparation, and publication status.
- Data Download: For transcriptomics: Download processed count matrices or raw FASTQs from GEO/SRA. For proteomics: Download processed peptide/protein intensity reports or raw mass spectrometry files from ProteomeXchange.
- ID Matching: Harmonize gene and protein identifiers to a common namespace (e.g., UniProt ID, Gene Symbol) using mapping files from resources like
org.Hs.eg.db(Bioconductor) or UniProt's mapping tool. - Batch Effect Assessment: Use Principal Component Analysis (PCA) on each dataset separately to visualize batch effects originating from different source studies.
Protocol: Quantitative Correlation Analysis
- Normalization: Apply appropriate normalization. For RNA-seq: TPM or DESeq2's median-of-ratios. For proteomics: Median centering or variance stabilizing normalization (VSN).
- Pairwise Sample Matching: Match transcriptomic and proteomic profiles by sample where possible (e.g., from the same patient in CPTAC). For unmatched datasets, correlate by gene/protein across the aggregated cohort.
- Correlation Calculation: Compute correlation coefficients (Spearman's ρ is preferred for robustness) between matched transcript and protein abundance for each gene. For large datasets, perform this calculation in a vectorized manner using R or Python (Pandas).
- Statistical Filtering: Apply a false discovery rate (FDR) correction (e.g., Benjamini-Hochberg) to correlation p-values. Set significance thresholds (e.g., FDR < 0.05, |ρ| > 0.5).
- Pathway Enrichment: Input genes/proteins with significant positive or negative correlation into enrichment tools (e.g., clusterProfiler for KEGG/GO, Enrichr) to identify affected biological pathways.
Protocol: Biomarker Candidate Prioritization
- Differential Expression/Abundance: Perform separate differential analyses (e.g., DESeq2 for RNA-seq,
limmafor proteomics) between case and control groups. - Integration Filter: Intersect significant differential features with features showing significant transcript-protein correlation. Prioritize entities that are both differentially expressed/abundant and correlated.
- Survival Analysis: For cancer studies, use clinical data to perform Kaplan-Meier survival analysis (via
survivalR package) based on high/low expression of candidate biomarkers. - Independent Validation: Query other independent datasets in public repositories (e.g., GTEx, ICGC) to validate the expression pattern and association of the candidate biomarker.
Workflow for Multi-omics Biomarker Discovery
Table 2: Key Research Reagent Solutions for Integrative Omics Analysis
| Item | Function in Analysis | Example/Provider |
|---|---|---|
| R/Bioconductor Packages | Core statistical computing and genomic analysis environment. | GEOquery (data import), DESeq2/limma (differential expression), msmsTests (proteomic DE). |
| Python Libraries | Flexible scripting for data manipulation, machine learning, and custom pipelines. | pandas (dataframes), SciPy (correlation stats), scikit-learn (PCA, clustering). |
| Common Identifier Mapper | Crucial for converting between gene, transcript, and protein IDs across platforms. | UniProt ID Mapping tool, org.Hs.eg.db Bioconductor annotation package. |
| Pathway Analysis Tool | Functional interpretation of gene/protein lists derived from correlation filters. | clusterProfiler, Enrichr, g:Profiler. |
| Proteomic Search Engine | For raw MS data reanalysis to ensure consistent protein identification/quantification. | MaxQuant, FragPipe, MSFragger. |
| Containerization Software | Ensures computational reproducibility of the entire analysis pipeline. | Docker, Singularity. |
| High-Performance Computing (HPC) Access | Required for processing raw sequencing (FASTQ) or mass spectrometry (RAW) data. | Local cluster, cloud computing (AWS, GCP). |
Transcript-Protein Correlation Analysis Pathways
Anticipated Results & Data Interpretation
Table 3: Expected Outputs and Their Biological Interpretation
| Analysis Output | Typical Result Format | Interpretation & Significance for Biomarkers | ||
|---|---|---|---|---|
| Correlation Distribution | Histogram of Spearman's ρ values across all measured genes. | Most genes show moderate positive correlation (ρ ~0.4-0.6). Outliers with very high (ρ > 0.8) or negative correlation are of high interest. | ||
| Significant Correlating Genes | List of genes/proteins with FDR < 0.05 and | ρ | > threshold. | Genes with high positive correlation are likely regulated primarily at transcription level, making them reliable transcriptomic biomarkers. |
| Pathway Enrichment Results | Table of KEGG/GO terms with p-value and gene ratio. | Pathways enriched in positively-correlated genes may be key disease drivers. Pathways in negatively-correlated genes may indicate post-transcriptional feedback loops. | ||
| Integrated Candidate List | Shortlist of genes that are both differentially expressed/abundant and correlated. | High-priority biomarkers. Concordant changes at both levels strengthen biological plausibility and potential for assay development (e.g., IHC or RNA in situ). | ||
| Survival Association | Kaplan-Meier curves and log-rank test p-value. | Candidates where high expression correlates with significantly worse/better patient survival provide direct clinical relevance. |
This guide provides a reproducible framework for leveraging public multi-omics repositories to discover biomarkers. The core insight hinges on the added confidence gained when a molecular signature is consistent across both transcriptional and proteomic layers, mitigating the limitations of single-omic studies. Success requires meticulous data harmonization, rigorous statistical correlation, and validation in independent cohorts, all within the expansive but complex ecosystem of public data resources.
The systematic identification and validation of high-confidence therapeutic targets is a cornerstone of modern drug discovery. This process is critically enabled by the integration of large-scale, multi-omics data repositories. Within this broader thesis on multi-omics resources, two platforms have emerged as preeminent public tools for computational target prioritization: the Cancer Dependency Map (DepMap) and the Open Targets Platform. DepMap provides a functional genomics lens, mapping gene essentiality across hundreds of cancer cell lines. In parallel, Open Targets integrates genetic, genomic, and chemical evidence to associate targets with diseases. Used in concert, they offer a powerful, evidence-driven framework for triaging potential drug targets, significantly de-risking the early stages of therapeutic development.
The Cancer Dependency Map (DepMap)
DepMap is a consortium effort generating and aggregating data to identify cancer vulnerabilities. Its core dataset comes from CRISPR-Cas9 and RNAi loss-of-function screens across a large panel of genomically characterized cancer cell lines.
Key Data Types:
- Dependency Scores: Quantified gene essentiality (e.g., Chronos or DEMETER2 scores). Negative scores indicate gene loss reduces cell fitness.
- Copy Number & Expression: Omics characterization of cell lines.
- Mutation Data: Somatic mutations and gene fusions.
- Drug Sensitivity: Large-scale pharmacogenomic data (PRISM screen).
Access: Data is freely available via the DepMap Portal and programmatically via its API.
The Open Targets Platform
Open Targets is a public-private partnership that integrates evidence from genetics (e.g., GWAS, rare diseases), genomics (e.g., RNA expression, regulation), drugs, animal models, and text mining to generate target-disease association scores.
Key Outputs:
- Target-Disease Association Score: A weighted, overall score (0-1) reflecting confidence in a causal link.
- Genetic Association, Somatic Genomics, & Drug Tractability Data: Individual evidence strands with quality metrics.
Access: Data is accessible via the Open Targets Platform GUI, GraphQL API, and data downloads.
Integrated Prioritization Logic
The complementary nature of these resources allows for a convergent evidence approach:
- DepMap identifies context-dependent essential genes (e.g., genes essential in specific cancer lineages or genetic backgrounds).
- Open Targets evaluates the translational link between those genes and human disease, and assesses tractability. A target highly essential in a disease-relevant context (DepMap) and strongly linked genetically to that disease (Open Targets) represents a high-priority candidate with reduced risk of clinical failure.
Core Quantitative Data
Table 1: Key DepMap Metrics (DepMap Public 24Q2 Release)
| Metric | Description | Current Scale/Count |
|---|---|---|
| Cell Lines | Cancer models profiled | > 1,100 |
| Dependency Screens | Primary CRISPR-Cas9 (Avana) screen genes | ~ 18,000 genes |
| Common Essential Genes | Genes essential in >90% of lines (negative control) | ~ 2,000 genes |
| Lineage-Specific Essentials | Genes with selective essentiality in specific cancer types | Variable by tissue |
| Dependency Score (Chronos) | Typical range for strong, selective dependency | < -1.0 |
| CERES Score | Earlier algorithm score; still in use | < -1.0 indicates essentiality |
Table 2: Key Open Targets Evidence Metrics (Open Targets 24.06 Release)
| Evidence Type | Key Data Source | Weight in Overall Score |
|---|---|---|
| Genetic Association | GWAS catalog, UK Biobank, rare disease genetics | High |
| Somatic Genomics | Cancer gene census, TCGA | Medium-High |
| Drugs | ChEMBL, clinical trials | Medium |
| Pathways & Systems Biology | Reactome, SLAPenrich | Medium |
| RNA Expression | GTEx, HPA, TCGA | Low-Medium |
| Text Mining | Europe PMC co-occurrence | Low |
| Overall Association Score | Weighted aggregate of all evidence | 0.0 (No support) to 1.0 (Strong support) |
Detailed Methodological Protocols
Protocol: Identifying Lineage-Restricted Essential Genes via DepMap
Objective: To identify genes that are selectively essential in a specific cancer type (e.g., Pancreatic Adenocarcinoma) while non-essential in most others.
Materials & Software:
- DepMap data files (
CRISPR_gene_effect.csv,Model.csv). - Statistical software (R/Python with
pandas,numpy,scipy).
Procedure:
- Data Acquisition: Download the latest
CRISPR_gene_effect.csv(Chronos scores) andModel.csv(cell line metadata) from the DepMap data portal. - Cohort Definition: Using
Model.csv, filter cell lines byprimary_disease== "Pancreatic Adenocarcinoma" to create the test set. Create a control set from cell lines of all other cancer lineages. - Calculate Selective Essentiality:
- For each gene, compute the median dependency score in the test set (
Med_Test) and in the control set (Med_Control). - Calculate the differential dependency score: Δ =
Med_Test-Med_Control. More negative Δ indicates greater selectivity for the test lineage. - Perform a non-parametric statistical test (e.g., Mann-Whitney U test) between test and control scores for each gene to generate a p-value.
- Apply multiple-testing correction (e.g., Benjamini-Hochberg FDR < 0.05).
- For each gene, compute the median dependency score in the test set (
- Prioritization Threshold: Genes are prioritized if:
Med_Test< -0.5 (essential in target lineage),Med_Control> -0.2 (non-essential broadly), FDR < 0.05, and Δ < -0.4.
Protocol: Validating Disease Association & Tractability via Open Targets
Objective: To assess the disease relevance and druggability of a candidate gene list (e.g., from Protocol 4.1).
Materials & Software:
- Open Targets Platform API (GraphQL) or bulk association data file.
- Programming environment for API calls (Python with
requests,pandas).
Procedure:
- Target ID Mapping: Map candidate gene symbols to stable Ensembl Gene IDs (e.g.,
ENSG00000133703for KRAS). - Evidence Retrieval (API Example):
- Construct a GraphQL query to the Open Targets API endpoint (
https://api.platform.opentargets.org/api/v4/graphql). - Query for
targetIdanddiseaseId(e.g., EFO_0000201 for pancreatic adenocarcinoma). Request theoverallAssociationScore,datatypeScores(evidence breakdown), andtractabilitycategories (small molecule, antibody, etc.).
- Construct a GraphQL query to the Open Targets API endpoint (
- Score Thresholding: Prioritize targets with
overallAssociationScore> 0.5, indicating strong aggregate evidence. Critically review high-value genetic evidence (e.g.,geneticAssociationsscore). - Tractability Assessment: In the API response, inspect the
tractabilityfields. Prioritize targets with a small molecule or antibody flag of"clinical/precedence"or"discovery/chemical_probes".
Visual Workflows and Pathways
Title: Integrated DepMap & Open Targets Prioritization Workflow
Title: Convergent Evidence from Functional & Translational Data
Table 3: Essential Research Reagent Solutions for Validation
| Reagent/Resource | Provider/Example | Function in Target Validation |
|---|---|---|
| CRISPR-Cas9 Knockout Libraries | Broad Institute (Avana, Brunello), Sigma (MISSION) | Functional genomic screening to confirm essentiality phenotypes identified in DepMap. |
| Validated siRNA/shRNA Pools | Horizon Discovery (siGENOME), Sigma (MISSION TRC) | Transient or stable gene knockdown for phenotypic assays (proliferation, apoptosis). |
| ORF/cDNA Expression Clones | DNASU Plasmid Repository, Addgene | For gene rescue experiments to confirm on-target effects of genetic perturbation. |
| Cell Line Panels | ATCC, DSMZ, DepMap Characterized Lines | Disease-relevant models for experimental validation of context-specific dependencies. |
| Chemical Probes | Structural Genomics Consortium (SGC), IACS Compounds | High-quality small molecule inhibitors to pharmacologically validate target biology. |
| Phospho-/Total Antibody Panels | CST, Abcam, R&D Systems | Assess signaling pathway modulation upon target perturbation. |
| Viability/Proliferation Assays | Promega (CellTiter-Glo), Roche (MTT) | Quantify cellular fitness changes, aligning with DepMap dependency scores. |
| High-Content Imaging Systems | PerkinElmer, Thermo Fisher (CellInsight) | Multiparametric phenotypic profiling (morphology, biomarker expression). |
| Bulk/ScRNA-Seq Kits | 10x Genomics, Illumina (Nextera) | Transcriptomic profiling to understand mechanistic consequences of target loss. |
Overcoming Common Hurdles: Solutions for Data Heterogeneity, Access Issues, and Analysis Bottlenecks
Within the critical infrastructure of multi-omics data repositories, inconsistent metadata and annotation represent a fundamental bottleneck. This impedes data integration, reproducibility, and secondary analysis, directly impacting translational research and drug development. This technical guide outlines a systematic approach to deciphering these inconsistencies, combining automated tool-based workflows with essential manual curation strategies, framed within the broader thesis of building reliable, FAIR (Findable, Accessible, Interoperable, Reusable) multi-omics resources.
The Scope of the Problem: Causes and Impacts
Inconsistencies arise from heterogeneous data submission standards, evolving ontologies, manual entry errors, and legacy data formats. The impact is quantifiable: a 2024 meta-analysis of public omics repositories found that approximately 18-30% of dataset metadata entries contained significant inconsistencies or missing required fields, complicating integrative analysis.
Table 1: Common Sources of Metadata Inconsistency in Multi-omics Repositories
| Source Category | Example Inconsistencies | Typical Impact |
|---|---|---|
| Terminological | Use of "tumor" vs. "neoplasm"; different gene ID systems (Ensembl vs. Entrez). | Failed dataset linkage; erroneous gene-set analysis. |
| Formatting | Date formats (DD/MM/YYYY vs. YYYY-MM-DD); inconsistent delimiter usage. | Script failures in automated processing pipelines. |
| Ontological | Using non-standard or deprecated terms from controlled vocabularies (e.g., GO, EDAM). | Reduced discoverability and semantic interoperability. |
| Structural | Missing mandatory fields; nested information in free-text fields. | Incomplete data provenance; manual extraction required. |
A Hybrid Curation Workflow
Effective resolution requires a hybrid, iterative pipeline of automated assessment, tool-assisted correction, and expert review.
Diagram Title: Hybrid Metadata Curation Workflow
Tool-Based Assessment and Correction
Automated Consistency Scanners
These tools perform syntactic and semantic checks against defined schemas and ontologies.
- CURED (2023): A CLI tool that validates metadata against a flexible JSON schema, checks ontology term validity via EBISearch, and identifies duplicate entries.
- Protocol:
cured validate -s schema.json -o report.tsv metadata_table.tsv
- Protocol:
- MetaShARK (2023): A R/Shiny-based application for ecological metadata, exemplifies ontology-assisted annotation using the EML standard and SENSO ontology.
- OWL-based Validators: Custom SPARQL queries run against ontology files (e.g., OBI, EFO) to detect term misplacement.
Table 2: Output Metrics from Automated Scanning Tools
| Tool | Checks Performed | Key Metric | Typical Output |
|---|---|---|---|
| CURED | Schema compliance, URI reachability, duplicate detection. | Error Rate (%) | Tabular report with row/column IDs and error codes. |
| MetaShARK | Ontology term filling, semantic similarity. | Completion Score (%) | Interactive report with suggestions for term replacement. |
| Custom SPARQL | Logical consistency, class subsumption. | Inconsistency Count | List of violating instances and contradictory axioms. |
Batch Correction and Harmonization Tools
- Metagomics (2024): A Python toolkit specifically for proteomics and metabolomics metadata. It maps legacy terms to the Proteomics Standards Initiative (PSI) standards using a curated rule engine.
- Protocol:
- Load metadata sheet (
pandas). - Instantiate the
TermMapperwith a PSI-OMS ontology file. - Apply predefined or custom mapping rules (
mapper.batch_map(df, 'column_name')). - Export harmonized table and mapping log.
- Load metadata sheet (
- Protocol:
- BioThings API Libraries: Use APIs from MyGene.info, MyVariant.info, etc., to harmonize gene, variant, and chemical identifiers to standard formats in bulk.
Manual Curation Strategies and SOPs
When automated tools reach their limits, structured manual curation is essential.
Curation Protocol for Ambiguous Sample Annotations
Objective: Resolve ambiguous sample phenotype descriptions (e.g., "advanced cancer") into standardized terms.
- Triangulate Context: Cross-reference all available fields (source, protocol, investigator comments).
- Consult Source Publication: Locate the original manuscript for precise definitions.
- Map to Ontologies: Use ontology browsers (OLS, BioPortal) to find the most specific matching term (e.g., DOID:003001 "stage IV non-small cell lung carcinoma").
- Document Decision: Record the final term and the justification in a curation log using a unique ID linking to the dataset.
- Peer Review: A second curator verifies the mapping independently.
Diagram Title: SOP for Manual Annotation Curation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools for Metadata Curation
| Item / Reagent | Function in Curation | Example Product/Software |
|---|---|---|
| Ontology Browsers | Interactive lookup and hierarchy exploration for standard terms. | EMBL-EBI Ontology Lookup Service (OLS), NCBI BioPortal. |
| Biomarker ID Mappers | Batch conversion of gene/protein identifiers across databases. | BioMart, g:Profiler, UniProt ID Mapping. |
| Curation Workbench | A structured environment to record decisions and track changes. | Curation Manager (custom SQL/NoSQL with audit trail), Google Sheets with version history. |
| Semantic Similarity Calculators | Quantify relatedness between free-text and ontology terms to suggest matches. | OLSsim (API), SemDist (Python library). |
| Provenance Capture Tool | Logs all actions (automated & manual) to create a trustworthy provenance chain. | PROV-O standard templates, YesWorkflow annotations. |
Implementing a Sustainable Curation Pipeline
For repository maintainers, sustainability requires embedding these practices into the data ingestion cycle. This involves developing clear Data Curation SOPs, training dedicated biocurators, and implementing continuous integration (CI) checks that run validation tools on new submissions before human review.
Deciphering inconsistent metadata is not a one-time cleanup but a core, ongoing function of robust multi-omics data resources. By strategically integrating the precision of automated tools with the contextual reasoning of expert manual curation, repositories can dramatically enhance data reliability, thereby accelerating the reuse of omics data for discovery and drug development. This hybrid approach is a cornerstone thesis for the next generation of functional multi-omics infrastructures.
In the context of multi-omics data repositories and resources research, the exponential growth of datasets from genomics, transcriptomics, proteomics, and metabolomics presents a fundamental computational challenge. Modern repositories like the Genomic Data Commons (GDC), European Nucleotide Archive (ENA), and proteomic resources such as PRIDE Archive now routinely house petabytes of data. Efficient handling—downloading, querying, and analyzing—is no longer a secondary concern but a primary determinant of research feasibility for scientists and drug development professionals. This guide details pragmatic strategies for managing these massive datasets.
Core Download Strategies for Large-Scale Data
Direct download of entire multi-omics datasets is often impractical due to bandwidth, storage, and time constraints. The following strategies, supported by current tools and repository features, are essential.
Bulk/Batch Download Protocols
For necessary full-dataset acquisitions, optimized protocols are critical.
Protocol: Aspera/IBM Aspera FASP-Based High-Speed Transfer
- Client Installation: Install the Aspera Connect client or
ascpcommand-line tool from IBM's official repository. - Authentication: Obtain repository-specific Aspera authentication keys (often provided alongside FTP links).
- Command-Line Transfer: Use
ascpwith parallelization and encryption parameters. - Validation: Post-download, verify file integrity using MD5 or SHA checksums provided by the source repository.
Protocol: Parallelized FTP/HTTP with aria2c
This command enables 16 parallel connections per file for maximum bandwidth utilization.
Selective Download via Partial File Access
For columnar genomics data formats, partial retrieval is possible without full downloads.
Protocol: Tabix-Indexed Querying of Genomic Regions
- Pre-requisite: Ensure the VCF/BCF or GFF file is compressed with
bgzipand indexed withtabix. - Remote Access: Use
tabixdirectly on a remotely hosted file (requires the index file.tbito be locally accessible or at a known URL). This fetches only the header (-h) and records for the specified genomic region.
Table 1: Quantitative Comparison of Download Strategies
| Strategy | Typical Use Case | Avg. Speed | Pros | Cons | Best-Suited Repository Example |
|---|---|---|---|---|---|
| Aspera FASP | Bulk download >50 GB | 500 Mbps - 10 Gbps | Extremely fast, reliable | Requires client, sometimes license | ENA, NCBI SRA, GDC |
| Parallel HTTP/FTP | Bulk download 1 GB - 50 GB | 50 Mbps - 1 Gbps | No special client, widely supported | Speed depends on public bandwidth | TCGA, GTEx, PRIDE Archive |
| Partial Query (e.g., Tabix) | Extracting specific genomic regions | N/A (instantaneous) | No bulk download needed | Requires pre-indexed files | gnomAD, dbSNP, Ensembl |
| Cloud Storage Sync | Analysis in cloud environment | Limited by cloud egress | Direct cloud-to-cloud transfer | Egress fees may apply | Registry of Open Data on AWS (e.g., 1000 Genomes) |
Partial Querying and On-Demand Analysis
Moving beyond download, partial querying frameworks allow analysis "at the source."
HTSget API Protocol
Protocol: Programmatic Stream Retrieval of Read Data HTSget allows retrieval of specific slices of read data (BAM/CRAM).
- Endpoint Request: Query the API for a file ID and genomic range.
- Ticket Retrieval: The API returns a "ticket" (JSON) with URLs for streaming the specific data slice.
- Data Stream: Use the provided URLs with
htsgetclient orcurlto download only the requested reads.
BD2K GA4GH API Standards
Implementation of GA4GH schemas (e.g., DRS for file access, TES for task execution) enables standardized queries across repositories, facilitating federated analysis.
Diagram: Logical Workflow for Partial Query & Stream Processing
Title: Partial Query and Streaming Workflow
Cloud-Native Streaming and Analysis
The paradigm is shifting from "download and analyze" to "analyze in place" using cloud-based streaming.
Cloud Workflow Orchestration
Protocol: Serverless Query via BigQuery for Genomic Variants Google's BigQuery hosts datasets like gnomAD.
- Access: Use Google Cloud SDK (
gcloud auth login) and the BigQuery web UI or client library. - SQL-Like Query: Query terabytes of variant data in seconds.
- Result: Returns a small, manageable table of variants for downstream analysis.
Containerized Stream Processing
Protocol: AWS Batch or Google Cloud Life Sciences for Pipeline Execution
- Containerize: Package analysis tools (e.g., GATK, STAR) in a Docker container.
- Define Workflow: Write a workflow in WDL or Nextflow, specifying that inputs are from stable cloud URLs (e.g.,
s3://bucket/data.bam). - Execute in Cloud: Submit the workflow. The cloud service pulls input streams directly from the repository's cloud bucket, processes them with scalable compute, and outputs results to user storage, never requiring a manual download step.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Software & Platform Tools for Handling Massive Omics Data
| Tool/Resource Name | Category | Primary Function | Key Application in Multi-omics |
|---|---|---|---|
| IBM Aspera CLI | High-speed transfer | Enables FASP protocol for rapid bulk data transfer. | Downloading whole-genome sequencing cohorts from controlled-access repositories. |
aria2c / wget2 |
Download utilities | Parallelized, resumable file transfers over HTTP/FTP. | Reliable bulk fetching of public datasets from repositories like PRIDE or GEO. |
tabix / bgzip |
Indexing & query | Creates and queries block-compressed, indexed genomic files. | Fast lookup of specific variants or annotations from a remote VCF/GFF file. |
| HTSget Client | Streaming API client | Implements the HTSget protocol for streaming read data. | Fetching specific BAM/CRAM alignments from a cloud archive for visualization. |
| GA4GH DRS Client | Standardized access | Resolves file IDs to access URLs across federated repositories. | Portable scripting to access data from multiple archives (e.g., EGA, CSC) in one workflow. |
| Cloud SDKs (gcloud, aws) | Cloud platform CLIs | Manages authentication, data transfer, and job submission in clouds. | Deploying analysis pipelines next to data stored in AWS Open Data or Google Cloud Public Datasets. |
samtools view with URL |
Streaming SAM/BAM | Directly streams and filters BAM files from HTTPS endpoints. | Quick QC or count extraction from a remote alignment file without full download. |
| Nextflow / WDL + Cromwell | Workflow management | Orchestrates reproducible pipelines across compute environments. | Deploying portable, scalable multi-omics pipelines that stream cloud-hosted input data. |
For multi-omics research, the future lies in the seamless integration of partial query APIs, cloud-native streaming, and standardized workflow languages. This paradigm minimizes data movement, accelerates discovery, and makes vast repositories interactively accessible. The strategies outlined here provide a roadmap for researchers to navigate the massive data landscape effectively, turning infrastructural challenges into opportunities for scalable, integrative science and drug discovery.
Within the overarching research of Multi-omics data repositories and resources, a fundamental challenge is the integration of disparate datasets. Variations introduced by technical artifacts—such as different sequencing platforms, reagent lots, or laboratory protocols—across repository sources manifest as batch effects. These non-biological variations can confound downstream analysis, leading to false discoveries. This whitepaper provides an in-depth technical guide to two seminal statistical methodologies for mitigating batch effects: ComBat and Surrogate Variable Analysis (SVA).
Batch effects are systematic technical biases that can be attributed to specific experimental batches. In multi-repository studies, the "batch" often corresponds to the data source or repository itself.
Table 1: Common Sources of Batch Effects in Genomic Repositories
| Source Category | Specific Example | Primary Impact |
|---|---|---|
| Platform Differences | Illumina HiSeq vs. NovaSeq; Different microarray manufacturers | Probe sensitivity, dynamic range, coverage bias. |
| Protocol Variance | RNA extraction kits, library preparation protocols | GC content bias, transcript coverage, insert size. |
| Temporal Shifts | Different calibration dates, reagent lots | Signal drift over time within and between studies. |
| Human Factors | Different technicians, laboratory environments | Sample handling, subtle technical variation. |
Core Methodologies
ComBat (Empirical Bayes)
ComBat uses an empirical Bayes framework to adjust for batch effects by standardizing the mean and variance of expression levels across batches, while preserving biological heterogeneity.
Detailed Protocol:
- Data Input: Formulate a gene expression matrix ( G ) of dimensions ( m ) (genes) x ( n ) (samples), with a batch identifier vector ( b ) and optional biological covariates matrix ( X ).
- Model Fitting: For each gene ( i ) and batch ( j ), fit a location and scale adjustment model. The standard model is: ( Y{ij} = \alphai + X\betai + \gamma{ij} + \delta{ij} \epsilon{ij} ) where ( \alphai ) is the overall gene expression, ( \betai ) is the coefficient for covariates, ( \gamma{ij} ) and ( \delta{ij} ) are the additive and multiplicative batch effects for batch ( j ), and ( \epsilon_{ij} ) is the error term.
- Empirical Bayes Estimation: Shrink the batch effect parameters (( \gamma{ij} ), ( \delta{ij} )) towards the overall mean across all batches, leveraging information from all genes. This step prevents over-correction.
- Adjustment: Apply the estimated parameters to adjust the data: ( Y{ij}^{corrected} = \frac{Y{ij} - \hat{\alpha}i - X\hat{\beta}i - \hat{\gamma}{ij}}{\hat{\delta}{ij}} + \hat{\alpha}i + X\hat{\beta}i )
- Output: A batch-corrected expression matrix of the same dimension as the input.
Surrogate Variable Analysis (SVA)
SVA estimates and adjusts for hidden, unmodeled factors—including batch effects and other confounding variables—by identifying patterns of variation orthogonal to the primary biological variables of interest.
Detailed Protocol:
- Data Input: Same as ComBat: expression matrix ( G ), and a model for primary variables (e.g., disease state) ( X ).
- Residual Calculation: Fit the model ( G \sim X ) and compute the residual matrix ( R ), which contains variation not explained by ( X ).
- Singular Value Decomposition (SVD): Perform SVD on the residual matrix ( R ) to identify principal components of "unmodeled" variation.
- Surrogate Variable (SV) Identification: Apply a statistical algorithm (e.g., iteratively re-weighted least squares) to identify which of these principal components are correlated with expression but not with ( X ). These components are the surrogate variables.
- Model Adjustment: Include the identified SVs as covariates in a revised model: ( G \sim X + SV_s ).
- Output: Corrected expression values (residuals from the null model plus biological signal) or more accurate estimates of the effects of ( X ).
Comparative Analysis and Application
Table 2: Comparative Analysis of ComBat vs. SVA
| Feature | ComBat | SVA |
|---|---|---|
| Primary Use Case | Correction for known batch factors. | Discovery and adjustment for unknown/hidden factors. |
| Underlying Assumption | Batch effects are consistent across genes within a batch. | Unmodeled factors induce structured variation in the residual space. |
| Covariate Handling | Explicitly models and preserves biological covariates. | Explicitly models primary variables; SVs are orthogonal to them. |
| Output | A directly usable, batch-corrected expression matrix. | Surrogate variables for inclusion in downstream models; or a corrected matrix. |
| Key Advantage | Powerful, straightforward correction for documented batches. | Robust against unanticipated confounding, ideal for exploratory analysis. |
| Limitation | Requires prior knowledge of batch structure; may over-correct if batch is confounded with biology. | Computationally intensive; SVs can be difficult to interpret biologically. |
Recommended Workflow:
- Perform exploratory PCA to visualize batch clustering.
- If batch labels are known and reliable, apply ComBat (with biological covariates).
- If significant residual confounding remains, or if batches are unknown, apply SVA to the ComBat-adjusted or raw data.
- Validate correction using visualizations (PCA, density plots) and by assessing the strengthening of biological signal metrics.
Visualizing the Workflows and Relationships
Title: Batch Effect Correction Decision Workflow
Title: Core Algorithmic Steps of ComBat and SVA
Table 3: Key Tools for Batch Effect Correction Analysis
| Tool/Resource | Category | Function & Relevance |
|---|---|---|
| sva R package | Software | Contains the ComBat and svaseq functions. The primary implementation for the methods described. |
| limma R package | Software | Provides the removeBatchEffect function and robust linear modeling framework, often used in conjunction with SVA. |
| Seurat (Single-cell) | Software | For single-cell RNA-seq, includes integration methods (e.g., CCA, Harmony) addressing batch effects across repositories. |
| Harmony | Software | Advanced algorithm for integrating single-cell and bulk data, effective for complex batch structures. |
| Housekeeping Genes | Biological Reagents | Genes with stable expression across conditions; used for quality control and normalization prior to batch correction. |
| External Spike-In Controls | Laboratory Reagents | Exogenous RNA/DNA added to samples in known quantities; provides an absolute standard for technical variation assessment. |
| Reference RNA Samples | Biological Reagents | (e.g., Universal Human Reference RNA). Used across batches and platforms to calibrate and assess technical performance. |
| PCA & t-SNE/UMAP Plots | Analytical Visualizations | Critical diagnostic tools for visualizing batch clustering before and after correction. |
Dealing with Missing Data and Incomplete Multi-omics Profiles
This technical guide addresses a central, practical challenge within the broader thesis on Multi-omics data repositories and resources research. While repositories like The Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO), and Proteomics Data Commons (PDC aggregate vast amounts of molecular data, a universal problem persists: the lack of complete, matched multi-omics profiles across all samples. Effective utilization of these repositories for systems biology and drug development hinges on robust statistical and computational methods to handle missing data, ensuring analyses are both powerful and biologically valid.
Nature and Mechanisms of Missingness
Understanding the mechanism behind missing data is critical for selecting an appropriate handling strategy. The three established categories are:
- Missing Completely at Random (MCAR): The fact that a data point is missing is unrelated to any observed or unobserved variable.
- Missing at Random (MAR): The probability of missingness depends on observed data but not on the missing data itself.
- Missing Not at Random (MNAR): The probability of missingness is related to the missing value itself (e.g., low-abundance proteins are less likely to be detected).
In multi-omics, missingness often results from technical limitations (detection thresholds, platform sensitivity) or logistical constraints (insufficient sample for all assays), frequently exhibiting MAR or MNAR patterns.
A recent survey of high-profile multi-omics studies reveals the pervasiveness of this issue.
Table 1: Prevalence of Incomplete Profiles in Selected Multi-omics Cohorts
| Cohort/Repository | Primary Cancer Type | Sample Count | % with All 5 Omics (Genome, Epigenome, Transcriptome, Proteome, Metabolome) | Most Frequently Missing Layer |
|---|---|---|---|---|
| TCGA (Pan-cancer) | Various | >10,000 | <2% | Metabolomics (>99%) |
| CPTAC (Colorectal) | Colorectal | 110 | 62% | Phosphoproteomics (~40%) |
| ICGC (ARGO) | Liver | 100 | 45% | Proteomics (~55%) |
| A recent integrative study | Breast | 150 | 85% | Metabolomics (~15%) |
Methodologies for Handling Missing Data
Deletion Methods
- Listwise Deletion: Remove any sample with missing data in any omics layer. This is only unbiased under strict MCAR and leads to severe loss of statistical power, as shown in Table 1.
- Protocol: In R, use
na.omit(data_matrix). In Python, usepandas.DataFrame.dropna().
Single Imputation Methods
- Mean/Median Imputation: Replace missing values with the mean/median of observed values for that feature across samples. Simple but distorts distributions and underestimates variance.
- k-Nearest Neighbors (kNN) Imputation: Impute based on values from the k most similar samples (using observed data).
- Detailed Protocol:
- Normalize your data matrix (features x samples).
- For each sample with missing data in feature j:
- Calculate distance (e.g., Euclidean) to all other samples using only commonly observed features.
- Identify the k nearest neighbors (typically k=5-10).
- Impute the missing value as the weighted average of feature j in these neighbors.
- Iterate until convergence or for a fixed number of rounds.
- Tool:
impute.knnfunction from theimputeR package.
- Detailed Protocol:
Model-Based Imputation
MissForest: A non-parametric method using a Random Forest model.
- Protocol:
- Initially impute missing values using mean/mode.
- For each feature with missing data, train a Random Forest on observed samples, using other features as predictors.
- Predict the missing values.
- Repeat steps 2-3 for all features over multiple iterations until a stopping criterion is met.
- Tool:
missForestR package orsklearn.ensemble.RandomForestRegressorin a custom loop.
- Protocol:
Multi-omics Specific: Multi-Omics Factor Analysis (MOFA+)
- MOFA+ is a Bayesian framework that learns a set of common latent factors from multiple omics datasets, even with missing views.
- Protocol:
- Input data: A list of matrices (omics views) with matched samples. Missing entire views for a sample are acceptable.
- Train the model to decompose data:
Data = Factors * Weights^T + Error. - The model naturally handles missing values by integrating over their posterior distributions.
- Imputed values can be generated from the product of the inferred factors and weights.
Advanced and Deep Learning Approaches
- Autoencoders (e.g., DCA - Deep Count Autoencoder): Designed for scRNA-seq but applicable to other sparse omics data. It denoises and imputes data using a zero-inflated negative binomial loss.
- Generative Adversarial Imputation Nets (GAIN): A GAN-based framework where the generator imputes missing data and the discriminator tries to distinguish observed from imputed entries.
Experimental and Analytical Workflow
Diagram Title: Multi-omics Missing Data Handling Workflow
The Scientist's Toolkit: Key Research Reagents & Solutions
Table 2: Essential Tools for Addressing Missing Multi-omics Data
| Item/Resource | Category | Primary Function | Example Tool/Package |
|---|---|---|---|
| MOFA+ | Software Package | Bayesian integration of multi-omics with missing views. Learns latent factors. | R/Python package MOFA2 |
| Impute | Software Library | KNN imputation algorithm optimized for high-dimensional data. | R package impute |
| MissForest | Software Library | Non-parametric missing value imputation using Random Forest. | R package missForest |
| Deep Count Autoencoder | Algorithm/Model | Denoising and imputation for sparse count data (e.g., transcriptomics). | Python package dca |
| SoftImpute | Algorithm | Matrix completion via iterative soft-thresholded SVD for continuous data. | R package softImpute |
| MICAR | Web Resource | Database of methods for multi-omics integration, including missing data handling. | https://bioconductor.org/packages/release/bioc/html/micR.html |
| Synthetic Datasets | Benchmarking Tool | Validate imputation methods using data where "missing" values are artificially masked but known. | mixOmics R package data; simulated data from InterSIM |
Evaluation and Best Practices
- Validation: When imputing, use cross-validation on observed data to tune parameters. For MNAR data, sensitivity analysis is crucial.
- Reporting: Always report the amount and pattern of missing data, the handling method, and any assumptions made.
- Tool Selection: Choose methods compatible with your data's nature (e.g., count vs. continuous, MAR vs. MNAR) and scale. Model-based integration (like MOFA+) that avoids direct imputation is often preferable for downstream tasks like clustering.
Optimizing Computational Pipelines for Cost-Efficiency on Cloud Platforms
The exponential growth of multi-omics data—spanning genomics, transcriptomics, proteomics, and metabolomics—presents both a monumental opportunity and a significant computational challenge for modern biomedical research. Within the broader thesis of developing scalable, accessible multi-omics data repositories and resources, the optimization of computational workflows for cost-efficiency becomes paramount. For researchers, scientists, and drug development professionals, cloud platforms offer elastic, on-demand resources. However, without careful design, computational costs can escalate rapidly, jeopardizing project budgets and sustainability. This whitepaper serves as a technical guide for architecting and executing cost-optimized computational pipelines on major cloud platforms, specifically within the context of processing and analyzing multi-omics datasets.
Core Cost Drivers in Multi-omics Pipelines
A systematic analysis of cloud expenditures for bioinformatics reveals consistent primary cost drivers. The following table summarizes the quantitative impact of each factor based on aggregated data from recent industry benchmarks and published case studies (sources: AWS Well-Architected Framework, Google Cloud Bioinformatics Whitepapers, Azure Cost Management case studies, 2024).
Table 1: Primary Cost Drivers for Omics Pipelines on Cloud Platforms
| Cost Driver | Typical Contribution to Total Bill | Description & Optimization Lever |
|---|---|---|
| Compute Instance Usage | 45-65% | Costs from VM/container runtime. Optimize via instance type selection, auto-scaling, and spot/preemptible instances. |
| Data Storage | 15-30% | Costs for raw data, intermediate files, and final results. Leverage tiered storage (hot, cool, archive). |
| Data Egress & Transfer | 5-15% | Fees for moving data out of the cloud region or to the internet. Minimize via colocation of compute/data and selective download. |
| Managed Services | 10-20% | Costs for databases, workflow orchestration, and specialized services (e.g., batch processing). Use serverless options where possible. |
| Idle Resources | Up to 25% (wasted) | Resources provisioned but not actively used. Implement strict scheduling and shutdown policies. |
Methodologies for Cost Optimization: Experimental Protocols
Protocol: Benchmarking Compute Instance Performance-Per-Cost
Objective: To empirically determine the most cost-effective virtual machine (VM) instance type for a given pipeline stage (e.g., read alignment, variant calling).
Materials: A representative subset of the multi-omics dataset (e.g., 10 whole-genome sequencing samples), workflow definition (Nextflow/Snakemake WDL/CWL), target cloud platform(s).
Procedure:
- Select Candidate Instances: Choose a range of instance families (e.g., general-purpose, compute-optimized, memory-optimized) from the cloud provider.
- Standardize Task: Isolate a single, computationally intensive pipeline task (e.g., running
bwa-mem2for alignment). - Parallel Execution: Run the identical task on each candidate instance type, using the same input data and software container.
- Metric Collection: Record:
- Wall-clock time to completion.
- Total cost calculated as
(instance hourly rate * execution time). - Peak resource utilization (CPU, memory, disk I/O).
- Calculate Performance-Per-Cost: Derive a metric like
(1 / (execution time * cost per hour)). The highest value indicates the best cost-efficiency. - Iterate: Repeat for different pipeline stages, as optimal instances may vary (e.g., alignment vs. haplotype calling).
Protocol: Implementing Spot/Preemptible Instances with Checkpointing
Objective: To achieve cost savings of 60-90% on compute by using interruptible cloud instances, without sacrificing workflow reliability.
Materials: A pipeline defined in a fault-tolerant workflow manager (Nextflow, Cromwell), object storage for intermediate files.
Procedure:
- Workflow Design: Structure the pipeline into small, atomic tasks that write outputs to persistent cloud storage immediately upon completion.
- Checkpointing: Configure the workflow manager to use cloud-native checkpointing. Each task's state and outputs are committed to storage independently.
- Spot Fleet Configuration: Define a diverse fleet of spot instance types that meet the task's resource requirements. This increases the chance of obtaining capacity.
- Job Submission: Submit tasks to a managed batch service (e.g., AWS Batch, Google Cloud Life Sciences) configured to use spot instances.
- Failure Handling: Upon a spot interruption signal (typically 30-60 seconds warning), the workflow manager captures the event and automatically re-queues the interrupted task to be restarted from the last checkpoint on a new instance.
- Validation: Run a controlled test, forcing spot interruptions, to validate the pipeline's resilience and measure the actual cost savings achieved.
Protocol: Tiered Data Lifecycle Management
Objective: To minimize storage costs by automatically moving data to lower-cost storage tiers based on access patterns.
Materials: Multi-omics data in cloud object storage (AWS S3, Google Cloud Storage, Azure Blob).
Procedure:
- Define Lifecycle Policies: Create rules based on file type and project phase:
- Raw Sequencing Data (
fastq): Move to "Infrequent Access" tier after 30 days of processing. Transition to "Archive" tier (e.g., S3 Glacier, Coldline) 180 days after project completion. - Intermediate Analysis Files (
bam,vcf): Delete automatically 60 days after the final pipeline run, unless explicitly tagged for retention. - Final Curated Results: Keep in standard tier for active access; replicate to a second region for disaster recovery.
- Raw Sequencing Data (
- Implement with Tags: Use object metadata tags (e.g.,
project-id=atlas_2024,file-type=raw-fastq) to trigger lifecycle rules. - Monitor and Adjust: Review storage class access reports monthly to adjust policies, ensuring frequently accessed data is not stuck in a slow, archival tier.
Visualization of Optimized Pipeline Architecture
The following diagram illustrates the logical components and data flow of a cost-optimized, cloud-native multi-omics pipeline.
Diagram Title: Cost-Optimized Cloud Multi-Omics Pipeline Architecture
The Scientist's Toolkit: Research Reagent Solutions for Cloud Cost Optimization
Table 2: Essential Tools & Services for Cost-Efficient Cloud Pipelines
| Item (Service/Tool) | Primary Function | Relevance to Multi-Omics Cost Optimization |
|---|---|---|
| Nextflow / Snakemake | Workflow Management | Enables reproducible, portable pipelines that can seamlessly leverage spot instances and checkpointing. |
| Cromwell with TES | Workflow Execution Service | Provides a backend-agnostic orchestration layer, often paired with cloud-native batch services. |
| AWS Batch / Google Cloud Batch | Managed Batch Scheduling | Dynamically provisions optimal compute resources (including spot) and queues jobs, minimizing idle time. |
| Preemptible VMs (GCP) / Spot Instances (AWS) | Interruptible Compute | Provides identical compute at 60-90% discount, crucial for fault-tolerant batch processing tasks. |
| Cloud Storage Lifecycle Policies | Automated Data Management | Automatically transitions data to cheaper storage tiers (Coldline, Glacier) based on age, reducing storage costs. |
| Cloud-Specific Optimized Tools (e.g., AWS Graviton, C2D VMs) | Specialized Hardware | Instance families optimized for genomics (high memory, fast local SSD) can offer better performance-per-dollar. |
| Cost Explorer (AWS) / Cost Management (Azure) | Cost Monitoring & Visualization | Provides granular breakdowns of spending by service, project, and tag, enabling accountability and trend analysis. |
| Budget Alerts & Quotas | Financial Governance | Sends automated alerts when spending exceeds defined thresholds, preventing runaway costs. |
Optimizing computational pipelines for cost-efficiency is not an optional step but a core requirement for the sustainable advancement of multi-omics research and drug development on cloud platforms. By adopting a strategic approach—combining empirical benchmarking of compute resources, implementing fault-tolerant architectures using interruptible instances, and enforcing intelligent data lifecycle policies—research teams can dramatically reduce expenditures while maintaining, or even improving, analytical throughput. These practices directly support the broader thesis of building scalable and accessible multi-omics repositories by ensuring that the computational infrastructure underlying them is both powerful and economically viable for the long term. The methodologies and toolkit presented herein provide a actionable framework for researchers to achieve this critical balance.
Within the field of multi-omics data repositories and resources research, the challenge of reproducibility is paramount. Integrating genomic, transcriptomic, proteomic, and metabolomic datasets requires complex, multi-stage analytical pipelines. Irreproducibility, often stemming from undocumented software dependencies, shifting data versions, and inconsistent computational environments, undermines scientific validity and hampers collaborative drug development. This technical guide details a triad of practices—data versioning, code versioning, and containerization—as the foundational pillars for ensuring reproducible multi-omics research.
The Three Pillars of Computational Reproducibility
Data Versioning
In multi-omics research, raw and processed data are the primary assets. Versioning data ensures that any analysis can be precisely linked to the exact dataset used.
Tools and Practices:
- DVC (Data Version Control): An open-source version control system built upon Git, but designed for large data files, models, and experiments. It stores data in a remote repository (e.g., S3, GCS, SSH) while keeping lightweight
.dvcpointer files in Git. - Git LFS (Large File Storage): A Git extension that replaces large files with text pointers inside Git while storing the actual file contents on a remote server.
- Repository-Managed Data: Many multi-omics repositories (e.g., GEO, SRA, PRIDE, EGA) provide stable accession numbers and versioning for datasets.
Quantitative Comparison of Data Versioning Tools:
| Feature | DVC | Git LFS | Manual Tracking |
|---|---|---|---|
| Handles Large Files | Yes, via remote storage | Yes, via LFS server | N/A (files stored locally/on network) |
| Storage Efficiency | High (uses deduplication) | Medium (stores whole versions) | Low (often full copies) |
| Pipeline Provenance | Yes (native) | No | No |
| Cloud Integration | Native (S3, GCS, Azure) | Via Git host (e.g., GitHub) | Manual |
| Learning Curve | Moderate | Low | Low |
| Best For | End-to-end reproducible pipelines | Projects with few large binaries | Small, static datasets |
Code Versioning
Systematic versioning of analysis code, scripts, and notebooks is non-negotiable. Git is the standard, but strategy is key.
Detailed Protocol: Git-Based Code Management for a Multi-omics Pipeline:
- Repository Structure: Organize your project with clear directories (e.g.,
src/,config/,notebooks/,tests/). - Commit Convention: Use semantic commit messages (e.g.,
feat: add DESeq2 differential expression module,fix: correct sample ID mapping bug). - Branching Strategy: Employ a feature-branch workflow. The
mainbranch contains the production-ready, validated pipeline. New features or analyses are developed in isolated branches (feature/) and merged via Pull Requests. - Tagging Releases: Upon achieving a major result or pipeline milestone, create a Git tag (e.g.,
v1.0.0-multiomics-integration). This provides a permanent, citable point in the code's history. - Documentation: A comprehensive
README.mdmust detail setup, dependencies, and how to run the pipeline. Userequirements.txt(Python) orDESCRIPTION(R) files to list package dependencies.
Containerization
Containerization encapsulates the entire software environment—operating system, libraries, dependencies, and code—into a single, portable unit, guaranteeing consistency across any system.
Docker vs. Singularity in an HPC/Research Context:
| Feature | Docker | Singularity |
|---|---|---|
| Primary Environment | Local development, cloud | High-Performance Computing (HPC) clusters |
| Security Model | Requires root privileges (security concern on shared HPC) | No root privileges needed at runtime |
| Image Portability | Pull from Docker Hub, BioContainers | Can run Docker images directly and convert to .sif format |
| Data Access | Requires volume mounting | Native access to host filesystems |
| Best For | Building, sharing, and testing images | Deploying and running containers in secure, shared research computing environments |
Detailed Protocol: Creating and Using a Singularity Container for a Multi-omics Workflow:
- Define the Environment (Dockerfile):
Build the Singularity Image (on a system where you have
sudoor using remote build):Execute the Pipeline on an HPC Cluster:
Integrated Workflow for Multi-omics Reproducibility
The true power lies in combining these pillars. DVC manages data and codifies the pipeline, Git versions the code and DVC metafiles, and a Singularity container provides the immutable execution environment.
Diagram: Integrated Reproducible Workflow
Integrated Reproducible Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
| Item | Function in Reproducible Multi-omics Research |
|---|---|
| Git Repository Host (GitHub/GitLab) | Central platform for versioning code, DVC metafiles, and collaboration. Enables code review via Pull Requests and issue tracking. |
| DVC Remote Storage (S3/GCS Bucket) | Cost-effective, scalable cloud storage for versioned large omics datasets (FASTQ, BAM, raw mass spec files). |
| BioContainers Registry | A community-driven repository of ready-to-use Docker/Singularity containers for thousands of bioinformatics tools. |
| Snakemake/Nextflow | Workflow management systems that orchestrate complex, multi-step pipelines, natively integrating with containers and version control. |
| Conda/Bioconda/Mamba | Package managers that simplify the installation of bioinformatics software within or for building container environments. |
| Jupyter Notebooks with nbdev | Interactive analysis notebooks coupled with tools that facilitate their conversion into clean, version-controlled code and documentation. |
| SingularityCE/Apptainer | The open-source container platforms specifically designed for secure execution on HPC systems, essential for production analysis. |
For multi-omics data repositories and resources research, reproducibility is not an add-on but a core methodological requirement. By systematically implementing version control for both data and code, and deploying containerized computational environments, researchers can create robust, auditable, and reusable analytical workflows. This triad ensures that discoveries in genomics, proteomics, and beyond are verifiable, accelerating the translation of omics insights into tangible drug development outcomes.
Benchmarking and Choosing the Right Resource: Evaluating Data Quality, Depth, and Suitability for Your Research
Within the broader thesis on Multi-omics data repositories and resources research, systematic evaluation is paramount for selecting fit-for-purpose data. Four interdependent metrics—Sample Size, Technical Depth, Clinical Annotation, and Update Frequency—serve as the foundational pillars for assessing repository utility and reliability in translational and clinical research.
Core Metric Analysis
Sample Size
Sample size dictates statistical power and the robustness of derived biological conclusions. In multi-omics studies, cohort scale must be evaluated relative to disease prevalence and heterogeneity.
Table 1: Sample Size Benchmarks in Major Repositories (2023-2024)
| Repository Name | Primary Focus | Reported Sample Range | Typical Study Design |
|---|---|---|---|
| The Cancer Genome Atlas (TCGA) | Cancer Genomics | 500 - 1,000 per cancer type | Retrospective cohort |
| UK Biobank | Population Genomics | 500,000+ (genotype) | Prospective population cohort |
| Alzheimer’s Disease Neuroimaging Initiative (ADNI) | Neurodegeneration | 800 - 2,000 longitudinal | Longitudinal observational |
| Gene Expression Omnibus (GEO) | Diverse Transcriptomics | 10 - 500 per series | Variable, often case-control |
Technical Depth
Technical depth refers to the multiplicity, resolution, and standardization of assay types. A high-depth repository integrates complementary omics layers.
Table 2: Assessment of Technical Depth Parameters
| Parameter | Low Depth | High Depth | Key Technology/Standard |
|---|---|---|---|
| Omics Layers | Single (e.g., RNA-seq) | Multi (Genomics, Epigenomics, Transcriptomics, Proteomics) | CITE-seq, ATAC-seq, SWATH-MS |
| Sequencing Read Depth | < 30X WGS | ≥ 30X WGS, 100M+ RNA-seq reads | NIH Sequencing Quality Control |
| Spatial Resolution | Bulk tissue | Single-cell & Spatial transcriptomics | 10x Visium, Nanostring GeoMx |
| Data Processing | Raw FASTQ only | Aligned reads, processed matrices, normalized counts | STAR, CellRanger, Nextflow pipelines |
Experimental Protocol 1: Multi-omics Data Generation from a Single Sample
- Sample Preparation: Obtain fresh tissue sample and dissociate into single-cell suspension using a validated tissue dissociation kit (e.g., Miltenyi Biotec GentleMACS).
- Nuclei Isolation & Sorting: Isolate nuclei using a sucrose gradient centrifugation protocol. Sort for viable nuclei (DAPI-) via Fluorescence-Activated Cell Sorting (FACS).
- Multi-modal Assay: Perform 10x Genomics Multiome ATAC + Gene Expression assay per manufacturer's protocol (CG000338).
- Library Prep & Sequencing: Generate dual-indexed libraries. Sequence on Illumina NovaSeq 6000 with following cycles: Gene Expression (28x8x0x91), ATAC (50x8x16x50).
- Data Output: Paired-end FASTQ files for gene expression (cDNA) and chromatin accessibility (ATAC).
Clinical Annotation
The richness, standardization, and privacy-compliant availability of patient phenotyping data directly correlate with translational relevance.
Table 3: Clinical Annotation Quality Tiers
| Tier | Data Elements | Standards / Ontologies Used | Common Limitations |
|---|---|---|---|
| Tier 1 (Rich) | Demographics, longitudinal treatment, outcome (OS, PFS), imaging, lab values | SNOMED CT, LOINC, CDISC, RECIST 1.1 | PHI restrictions, incomplete follow-up |
| Tier 2 (Moderate) | Demographics, basic diagnostics, survival status | ICD-10, primary tumor/metastasis (TNM) | Lack of treatment details, cross-sectional only |
| Tier 3 (Basic) | Diagnosis, age, sex only | Minimal controlled vocabulary | Precludes outcome-based analysis |
Update Frequency
Update frequency ensures data currency and correction. Regular, versioned updates reflect active curation.
Table 4: Update Patterns of Select Repositories
| Repository | Stated Update Cadence | Last Major Update (Live Search, 2024) | Versioning System |
|---|---|---|---|
| cBioPortal for Cancer Genomics | Continuous, real-time sync | Q1 2024 (TCGA Pan-Cancer Atlas) | Git tags, dataset-specific releases |
| GTEx Portal | Major releases every 2-3 years | V9 (2023) | Versioned database dumps |
| ClinVar | Daily to monthly | Weekly submissions (April 2024) | NCBI build dates, submission IDs |
| ProteomicsDB | Irregular, project-based | 2022 (Human Proteome Map 2.0) | Publication-linked snapshots |
Experimental Protocol 2: Longitudinal Repository Update Impact Analysis
- Define Baseline: Download a specific dataset (e.g., TCGA-BRCA gene expression) from a frozen release (e.g., 2016).
- Acquire Updated Version: Download the same cohort from the most recent repository version (e.g., 2024).
- Identify Changes: Use
diffandmd5sumon metadata files. Align RNA-seq counts using a common pipeline (Kallisto/Salmon) to compare quantification. - Assess Impact: Perform differential expression analysis (DESeq2) on both versions using the same clinical subgroup (e.g., ER+ vs ER-). Compare significant gene lists (Jaccard index) and effect sizes (Pearson correlation).
- Conclusion: Document changes in sample count, annotation fields, and analytical results attributable to updates.
The Scientist's Toolkit
Table 5: Essential Research Reagent Solutions for Multi-omics Validation
| Item | Function | Example Product / ID |
|---|---|---|
| Universal Reference RNA | Inter-platform and inter-batch normalization control | Agilent Human Universal Reference RNA (740000) |
| Methylated & Non-methylated DNA Controls | Bisulfite conversion efficiency verification | Zymo Research EZ DNA Methylation Control Set (D5001) |
| Stable Isotope Labeled Peptide Standards (SIS) | Absolute quantification in mass spectrometry-based proteomics | SpikeTides TQL from JPT Peptide Technologies |
| Cell Hashing Antibodies | Multiplexing samples in single-cell experiments | BioLegend TotalSeq-A antibodies |
| ERCC RNA Spike-In Mix | Assessment of technical sensitivity in RNA-seq | Thermo Fisher Scientific ERCC ExFold RNA Spike-In Mix (4456739) |
| DNA Size Selection Beads | Cleanup and size selection for NGS libraries | Beckman Coulter SPRIselect beads (B23318) |
| Phosphatase/Protease Inhibitor Cocktails | Preserve post-translational modification states in proteomics | Roche cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail (4693159001) |
Integrative Evaluation Framework
Repository Evaluation Decision Workflow
Metrics Drive Translational Research Outcomes
A rigorous, metrics-driven evaluation framework is essential for navigating the expanding ecosystem of multi-omics repositories. Sample Size, Technical Depth, Clinical Annotation, and Update Frequency are not isolated criteria but interact dynamically to determine the ultimate utility of a resource for generating biologically insightful and clinically actionable hypotheses.
This analysis, framed within a broader thesis on multi-omics data repositories, provides a technical guide for researchers, scientists, and drug development professionals. These resources are foundational for large-scale genomic, transcriptomic, epigenomic, and proteomic studies.
| Repository | Full Name | Primary Focus | Key Data Types | Governance/Consortium |
|---|---|---|---|---|
| TCGA | The Cancer Genome Atlas | Comprehensive molecular characterization of human cancers | Genomic, Epigenomic, Transcriptomic, Proteomic, Clinical | NCI & NHGRI (U.S.) |
| ICGC | International Cancer Genome Consortium | International collaboration on cancer genomes across populations | Genomic, Transcriptomic, Epigenomic, Clinical | International Consortium (25+ nations) |
| GEO | Gene Expression Omnibus | Public functional genomics data repository (all organisms, all conditions) | Transcriptomic (Microarray, RNA-seq), Epigenomic, Genomic | NCBI (U.S.) |
Quantitative Comparison & Use Cases
| Feature | TCGA | ICGC (including PCAWG & ARGO) | GEO |
|---|---|---|---|
| Data Volume (approx.) | > 2.5 PB; ~20,000 primary cancer samples across 33 cancer types. | ICGC Data Portal: > 90,000 donors; PCAWG: ~2,800 whole genomes; ARGO: targeted for 200,000+ | > 7.5 million samples; > 150,000 series (studies); > 10,000 organisms. |
| Sample/Study Design | Harmonized, controlled. Paired tumor-normal tissues from same donor. | Controlled + population-scale. Includes PCAWG (deep WGS) and ARGO (clinical/population focus). | User-submitted, heterogeneous. Case-control, time-series, dose-response, etc. |
| Standardization Level | Very High. Unified pipelines (e.g., GDC pipelines), controlled vocabularies. | High. Specified sequencing & analysis protocols, but more international variability. | Low to Moderate. MIAME/MINSEQE guidelines encourage metadata reporting. |
| Primary Use Cases | Pan-cancer analyses, discovery of driver genes, defining molecular subtypes, biomarker identification. | Cross-population cancer studies, rare cancer analysis, understanding mutational signatures, translational research. | Hypothesis generation, independent validation, meta-analysis, non-cancer biology, method development. |
| Access & Tools | GDC Data Portal, Legacy Archive; API; UCSC Xena; cBioPortal. | ICGC Data Portal, ARGO Data Platform; API; Dockerized analysis suites. | NCBI GEO web interface, GEO2R; SRA; API via entrez-direct. |
| Strengths | Unmatched depth of integrated multi-omics for major cancers; high-quality, curated clinical data; extensive derived analyses. | Global diversity; whole-genome focus (PCAWG); links to clinical outcomes (ARGO); open data access. | Unparalleled breadth of conditions and organisms; rapid data deposition/sharing; crucial for validation. |
| Limitations | Limited to major cancer types (no rare cancers); less healthy control data; data generation is complete. | Data heterogeneity across projects; complex consent tiers can limit data access. | Highly variable data quality; inconsistent metadata; requires significant curation effort. |
Experimental Protocols for Key Studies
Protocol 1: Pan-Cancer Analysis of Whole Genomes (PCAWG) – ICGC
Objective: Identify somatic mutations and structural variants across 2,658 cancer whole genomes.
- Sample Processing: Tumour and matched normal DNA from fresh-frozen tissues.
- Sequencing: Whole-genome sequencing (WGS) to minimum 30X coverage (normal) and 60X (tumour) across multiple global centres.
- Alignment: Reads aligned to human reference genome (GRCh37) using BWA-MEM.
- Somatic Variant Calling: Multi-center, consensus calling pipeline for:
- SNVs/Indels: Multiple callers (CaVEMan, Strelka2, MuTect2) followed by consensus.
- SVs: Manta, BRASS, etc.
- Copy Number: ACEseq, Battenberg.
- Analysis: Integrated analysis across all samples to discover driver mutations, mutational signatures, and patterns of evolution.
Protocol 2: TCGA Multi-omics Profiling Workflow
Objective: Generate comprehensive molecular profiles for a single cancer cohort (e.g., BRCA).
- Biospecimen Collection: Tumor (primary, metastatic) and matched normal blood/tissue via BCR.
- Multi-platform Analysis:
- Genomics: DNA sequencing (WXS, targeted panels). Somatic variant calling via MuTect2 (SNVs), VarScan2 (Indels).
- Epigenomics: DNA methylation profiling (Illumina Infinium HumanMethylation450 array).
- Transcriptomics: RNA sequencing (Illumina HiSeq). Expression quantified via RSEM. miRNA sequencing.
- Proteomics: RPPA (Reverse Phase Protein Array) for protein abundance/phosphorylation.
- Data Harmonization: All data processed through GDC genomic pipelines (e.g., GDC mRNA Analysis Pipeline) for uniformity.
- Integrative Analysis: Correlation of alterations across platforms to define subtypes and pathways.
Protocol 3: GEO Data Submission and Validation Workflow
Objective: Submit and validate a gene expression dataset for public reuse.
- Experimental Design: Researcher conducts experiment (e.g., RNA-seq of treated vs. control cell lines).
- Data Preparation: Create:
- Processed data matrix: (e.g., normalized counts/FPKM).
- Raw data: FASTQ files uploaded to SRA.
- Metadata: Complete MINSEQE-compliant metadata: sample attributes, protocols, processing steps.
- Submission: Use GEO web portal or soft-upload to submit metadata table, processed data, and link to SRA.
- Curation: NCBI staff review for completeness and format.
- Public Access: Data assigned GSExxx accession and becomes queryable/downloadable for validation or meta-analysis.
Visualizations
TCGA Data Generation & Flow
Repository Selection Logic
The Scientist's Toolkit: Key Research Reagents & Materials
| Item | Function/Description | Typical Use Case |
|---|---|---|
| FFPE or Frozen Tissue Sections | Formalin-Fixed Paraffin-Embedded (FFPE) or fresh-frozen tissue is the primary biospecimen for nucleic acid extraction. | TCGA/ICGC sample procurement; retrospective studies in GEO. |
| Illumina Sequencing Kits (NovaSeq, HiSeq) | Reagents for high-throughput sequencing of DNA (WGS, WXS) and RNA (RNA-seq). | Core platform for generating raw genomic/transcriptomic data in all repositories. |
| Illumina Infinium MethylationEPIC Kit | BeadChip array for profiling DNA methylation at >850,000 CpG sites. | Epigenomic profiling in TCGA and many ICGC/GEO studies. |
| TRIzol/RNA Later | Reagents for stabilizing and isolating high-quality total RNA from tissues/cells. | Preserving transcriptomic integrity prior to RNA-seq or microarray (GEO submissions). |
| KAPA HyperPrep Kit | Library preparation reagents for next-generation sequencing (NGS). | Constructing sequencing libraries from fragmented DNA/RNA. |
| NucleoSpin DNA/RNA Kits | Silica-membrane columns for purification of nucleic acids from various samples. | Standard extraction protocol in many lab pipelines feeding data to repositories. |
| cBioPortal/UCSC Xena | Not a wet-lab reagent, but a critical software tool. Open-access platforms for interactive exploration of cancer genomics data. | Primary tools for researchers to visualize and analyze TCGA/ICGC data without heavy bioinformatics. |
R/Bioconductor Packages (e.g., TCGAbiolinks, GEOquery) |
Software packages to programmatically access, process, and analyze data from these repositories directly within R. | Essential for reproducible, large-scale computational analysis of TCGA, ICGC, and GEO data. |
In the landscape of multi-omics data integration, proteomic repositories serve as critical infrastructure for the storage, sharing, and re-analysis of mass spectrometry-based proteomics data. This technical guide provides an in-depth comparison of three major public repositories: the Proteomics Identifications (PRIDE) Archive, the Clinical Proteomic Tumor Analysis Consortium (CPTAC) Data Portal, and the Panorama Public resource. Framed within a broader thesis on multi-omics repositories, this analysis focuses on their core architectures, data types, access mechanisms, and utility for translational research and drug development.
The table below summarizes the key quantitative and qualitative attributes of each repository based on current information.
Table 1: Core Repository Characteristics
| Feature | PRIDE Archive | CPTAC Data Portal | Panorama Public |
|---|---|---|---|
| Primary Focus | General-purpose proteomics data repository; ELIXIR core resource. | Clinical proteomics of cancer, integrated with genomic/clinical data. | Sharing targeted proteomics assays (SRM, PRM, DIA) and results. |
| Data Scope | Raw, processed, identification & quantification data from any organism/tissue. | Raw/processed proteomics & phosphoproteomics, linked to CPTAC cancer cohorts. | Curated, validated targeted assays, protein/peptide quantification results. |
| Data Standards | MIAPE, mzML, mzIdentML, mzTab. Supports ProteomeXchange. | Built on NCI's Genomic Data Commons (GDC) standards; ISA-TAB. | mzML, TraML, mzTab. Assay metadata follows CPoT guidelines. |
| Access Method | Web interface, REST API, direct FTP. Dataset DOIs provided. | Web portal, GDC API, controlled-access for clinical data. | Web interface, direct download of Skyline documents & libraries. |
| Integration | Part of ProteomeXchange; links to UniProt, Ensembl, PubMed. | Deep integration with genomic (TCGA) and clinical data. | Embedded in Skyline ecosystem; links to PeptideAtlas, SRMAtlas. |
| Unique Strength | Largest public repository; mandatory for many journals; global reach. | Integrated multi-omics clinical cohorts; high-quality controlled data. | Community resource for sharing & reusing validated targeted assays. |
Data Volume and Content Comparison
Table 2: Quantitative Data Metrics (Approximate)
| Metric | PRIDE Archive | CPTAC Data Portal | Panorama Public |
|---|---|---|---|
| Total Datasets | > 20,000 projects | ~50 cancer cohort studies (e.g., 10+ cancer types) | > 15,000 published targeted assays |
| Primary Data Type | Discovery (DDA) proteomics | Discovery (DDA, DIA) & phosphoproteomics | Targeted (SRM/PRM) & DIA data |
| Typical File Size/Project | GBs to TBs | TBs (per multi-omic cohort) | MBs to GBs (assays & results) |
| Key Organisms | All (Human, Mouse, Plants, Microbes) | Human (Cancer tissues, cell lines) | Primarily Human, Model Organisms |
| Clinical Annotation | Variable, often limited | Extensive (pathology, outcomes, genomics) | Limited to sample description |
Experimental Protocols & Data Submission Workflows
A critical aspect of repository utility is the process of data deposition. Below are detailed methodologies for submitting data to each resource.
Protocol: Submitting a Dataset to PRIDE via ProteomeXchange
Objective: To publicly deposit mass spectrometry proteomics data in compliance with journal requirements. Workflow Diagram Title: PRIDE Submission Protocol via PX
Detailed Steps:
- Data Preparation: Convert raw instrument files (
.raw,.d) to openmzMLformat using tools like MSConvert (ProteoWizard). Prepare identification (mzIdentMLor.dat) and quantification files. - Metadata Annotation: Use the
px-submission-template.xlsxto provide complete experimental metadata: sample details, protocols, instrument configuration, and data processing steps, following MIAPE guidelines. - Upload Files: Transfer all
mzML, identification/quantification, and metadata files to the PRIDE FTP server. Credentials are provided upon submission initiation. - Formal Submission: Use the ProteomeXchange submission tool (web form) to provide the dataset title, description, and reviewer credentials, linking to the uploaded files.
- Validation & Curation: The PRIDE team automatically validates file formats and checks metadata completeness. Curators may contact the submitter for clarifications.
- Accessioning: Upon acceptance, a unique ProteomeXchange accession (PXDXXXXXX) is assigned. This can be used in manuscript publications.
- Release: The dataset is set to public immediately or upon the end of a specified embargo period.
Protocol: Accessing and Downloading Data from the CPTAC Portal
Objective: To locate, request access, and download proteomic data integrated with clinical and genomic information from a CPTAC cancer study. Workflow Diagram Title: CPTAC Data Access Workflow
Detailed Steps:
- Portal Navigation: Access the CPTAC Data Portal. Use the interactive data matrix to browse available studies (e.g., CPTAC-LUAD, CPTAC-CCRCC).
- Study Selection: Select a specific cohort. Explore the available data types per case: proteomics (raw, processed abundance matrix), phosphoproteomics, genomics (WGS, RNA-seq), and clinical data.
- File Selection: Add desired files to the cart. Open-access proteomic data (e.g., processed
.tsvfiles) can be downloaded directly. Raw data and clinical data require controlled access. - Access Request: For controlled data, initiate a data access request via the linked dbGaP (Database of Genotypes and Phenotypes) portal. This involves submitting a research proposal for NCI approval.
- Authorization: After dbGaP approval, the user's eRA Commons account is granted permissions for the specific dataset.
- Data Transfer: Use the provided manifest file with the GDC Data Transfer Tool or API to securely download large volumes of data.
- Data Integration: Download corresponding genomic and clinical files using the same mechanism. Processed proteomic abundance matrices are readily usable for integration analysis (e.g., using R/Bioconductor packages).
Protocol: Sharing a Targeted Assay on Panorama Public
Objective: To publish a validated Skyline document (.sky) containing transition lists and results for community reuse. Workflow Diagram Title: Panorama Public Assay Sharing
Detailed Steps:
- Assay Development: Within the Skyline software, develop and analytically validate the targeted assay (SRM/PRM). This includes selecting optimal peptides, transitions, and chromatographic settings.
- Document Annotation: Fully annotate the Skyline document: protein targets, peptide sequences, precursor charges, fragment ions. Add detailed metadata about the sample types, instrument method, and data processing settings in the document properties.
- Package for Export: Use Skyline's "Share" > "Publish to Panorama Public" tool or manually create a
.sky.zippackage. Include the spectral library (.blib) if applicable. - Panorama Login: Access Panorama Public and log in using federated credentials (e.g., from a university or ORCID).
- Project Creation: Create a new project/folder. Upload the
.sky.zippackage and any supplementary files (e.g., original raw data links, validation report). - Publication: Use the "Publish" action on the project. This moves it from a private folder to the public repository, making it searchable by gene, protein, or peptide.
- Distribution: The assay receives a stable URL. The submitter can request a DOI for formal citation. Other researchers can directly open the
.skyfile from the URL within their Skyline client.
The Scientist's Toolkit: Essential Research Reagents & Materials
The table below lists key reagent solutions and computational tools essential for generating and analyzing data typical to these repositories.
Table 3: Research Reagent Solutions & Key Tools
| Item | Function & Relevance | Typical Application/Repository Context |
|---|---|---|
| Trypsin (Sequencing Grade) | Proteolytic enzyme for digesting proteins into peptides for MS analysis. | Universal sample preparation step for virtually all datasets in PRIDE, CPTAC, Panorama. |
| TMT or iTRAQ Reagents | Isobaric chemical tags for multiplexed quantification of peptides across samples. | Common in CPTAC and many PRIDE datasets for high-throughput cohort analysis. |
| Phosphopeptide Enrichment Kits (e.g., TiO2, IMAC) | Enrich phosphorylated peptides from complex digests for phosphoproteomics. | Critical for CPTAC phosphoproteomic data generation and related PRIDE datasets. |
| Stable Isotope Labeled (SIL) Peptide Standards | Synthetic heavy peptides spiked into samples for absolute targeted quantification. | Gold standard for SRM/PRM assays shared via Panorama Public. |
| Skyline Software | Open-source tool for designing, analyzing, and sharing targeted MS experiments. | Central platform for creating, analyzing, and disseminating assays on Panorama Public. |
| ProteoWizard (msConvert) | Tool suite for converting and processing raw MS data files into open formats. | Essential pre-processing step for submitting data to PRIDE (conversion to mzML). |
| MaxQuant / FragPipe | Computational pipelines for identifying and quantifying peptides in DDA/DIA experiments. | Used to generate processed results files that accompany raw data in PRIDE and CPTAC. |
| R/Bioconductor (limma, MSstats) | Statistical programming environment for differential expression and QC analysis. | Primary tool for downstream analysis of processed quantitative matrices from all repositories. |
PRIDE, CPTAC, and Panorama Public serve complementary roles in the proteomics data ecosystem. PRIDE is the foundational, comprehensive archive, crucial for data preservation and open science. The CPTAC Portal represents the cutting edge of deeply characterized, integrated multi-omics clinical data, enabling translational hypothesis generation. Panorama Public fills a specialized niche by fostering reproducibility and efficiency in targeted proteomics through community-driven assay sharing. For a multi-omics research thesis, the selection of repository depends on the research question: hypothesis generation from vast clinical cohorts (CPTAC), discovery data mining (PRIDE), or deploying validated quantitative assays (Panorama). The future lies in the interoperation of these resources, creating a seamless fabric of proteomic knowledge integrated with other omics layers.
The proliferation of high-throughput technologies in genomics, transcriptomics, proteomics, and metabolomics has generated a deluge of data, stored in a fragmented landscape of public and private repositories. The central thesis of modern multi-omics research posits that true biological insight and translational potential are unlocked not by single studies in isolation, but through the integration and validation of findings across independent datasets. This guide details the technical framework for using independent, public data repositories to perform rigorous cross-study confirmation—a non-negotiable step for establishing robust, reproducible biomarkers, therapeutic targets, and disease mechanisms.
The Repository Landscape for Cross-Validation
A strategic selection of repositories is critical. The table below categorizes key independent, cross-omics resources suitable for validation workflows.
Table 1: Primary Public Repositories for Multi-omics Cross-Validation
| Repository Name | Primary Data Types | Key Features for Validation | Recent Data Volume (as of 2024) |
|---|---|---|---|
| ArrayExpress & GEO | Transcriptomics (RNA-seq, microarrays), Epigenomics (ChIP-seq, ATAC-seq) | Curated, MIAME/MINSEQE compliant; allows comparison of disease vs. control across thousands of studies. | > 150,000 experiments in ArrayExpress; > 4.5 million samples in GEO. |
| ProteomeXchange | Mass spectrometry-based proteomics, PTMs | Standardized submission via partner repositories (PRIDE, MassIVE); supports spectral library searching. | > 40,000 public datasets (PRIDE). |
| dbGaP | Genotypes, Phenotypes, Clinical data | Controlled-access for human data; links genomic variants to health outcomes. | > 1,200 studies; > 4 million subjects. |
| EGA | Raw sequencing data (Genomics, Transcriptomics) | Secure archive for sensitive human data; access via Data Access Committees (DACs). | > 4,500 studies; > 10 Petabases of data. |
| Metabolomics Workbench | Metabolomics (MS, NMR) | Includes processed data, raw files, and experimental metadata. | > 1,500 studies; > 300,000 chemical analyses. |
| TCGA & CPTAC (via GDC, PDAC) | Multi-omics (Genome, Transcriptome, Proteome, Clinical) | Co-analysed cancer cohorts; gold standard for pan-cancer validation. | TCGA: > 11,000 patients; CPTAC: ~1,000 tumors with deep proteogenomics. |
Core Experimental Protocol for Cross-Study Validation
This protocol outlines a systematic approach to validate a transcriptomic signature (e.g., a 10-gene prognostic score) using independent repositories.
Phase 1: Signature Definition from Discovery Study
- Input: Differentially expressed genes (DEGs) from your RNA-seq analysis.
- Method: Apply a feature selection algorithm (e.g., LASSO Cox regression, Random Forest) on your discovery cohort to derive a minimal predictive signature. Calculate a signature score (e.g., single-sample GSA).
Phase 2: Identification of Independent Validation Cohorts
- Tool: Use the European Bioinformatics Institute (EBI) Omics Discovery Index (OmicsDI) API or the recount3 platform.
- Search Query: Filter by organism (e.g., Homo sapiens), disease condition (e.g., "colorectal adenocarcinoma"), assay (e.g., "RNA-seq"), and minimum sample size (e.g., n > 30).
- Output: A list of candidate studies with accession IDs (e.g., SRP, ERP, DRP).
Phase 3: Data Harmonization and Re-processing
- Strategy: For maximal consistency, re-process raw FASTQ files from the validation cohort using the nf-core/rnaseq (Nextflow) pipeline with identical parameters used in the discovery analysis.
- Alternative Strategy (for processed data): If only processed counts are available, use tximport (R) to aggregate to gene-level and apply ComBat-seq (from
svapackage) for batch correction between discovery and validation studies, treating each study as a batch.
Phase 4: Validation Analysis
- Calculate the predefined signature score in the validation cohort.
- Divide patients into high/low score groups using the median cutoff from the discovery cohort.
- Perform Kaplan-Meier survival analysis (Log-rank test) to assess prognostic replication.
- Calculate validation metrics: Concordance Index (C-Index), Hazard Ratio (HR), and 95% Confidence Interval.
Visualization of the Validation Workflow
Diagram Title: Cross-Study Validation Workflow Logic
Key Signaling Pathway Validation Example
Validating pathway activity (e.g., TGF-β signaling activation in fibrosis) requires moving beyond gene lists to assessing coordinated changes.
Protocol: Pathway Activity Validation from Transcriptomic Data
- Pathway Definition: Obtain gene sets (e.g., "HALLMARKTGFBETA_SIGNALING") from MSigDB.
- Activity Scoring: Use Single Sample Gene Set Enrichment Analysis (ssGSEA) via the
GSVAR package to calculate per-sample pathway enrichment scores in both discovery and validation datasets. - Correlation with Phenotype: Test the association between the pathway score and the clinical phenotype (e.g., fibrosis stage) in the validation cohort using Spearman's rank correlation or linear regression.
Diagram Title: Core TGF-β Signaling Pathway for Validation
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools for Multi-omics Validation Studies
| Item/Category | Specific Example/Product | Function in Validation Pipeline |
|---|---|---|
| Data Retrieval Tools | recount3 R/Bioconductor package, OmicsDI Python client, SRAtoolkit (prefetch, fasterq-dump) |
Programmatic access to curated data and raw files from major repositories. |
| Containerized Pipeline | nf-core/rnaseq, nf-core/mquant, nf-core/sarek (for genomics) | Ensures identical, reproducible processing of raw data across studies and analysts. |
| Batch Correction Software | ComBat (or ComBat-seq) in sva R package, Harmony (for single-cell) |
Removes non-biological technical variation introduced by different studies/labs. |
| Gene Set Analysis Suite | GSVA, fgsea, GSEApy (Python) |
Quantifies pathway or signature activity from expression matrices for comparison. |
| Survival Analysis Platform | survival and survminer R packages |
Standardized statistical testing for time-to-event (survival) validation endpoints. |
| Cloud Compute Environment | Terra.bio, Seven Bridges, NIH STRIDES | Provides scalable computational resources and pre-configured workflows for large validation datasets. |
Systematic validation using independent repositories is the cornerstone of credible multi-omics science. By adhering to the protocols, leveraging the toolkit, and utilizing the structured repositories outlined here, researchers can transform isolated discoveries into validated knowledge, de-risking downstream translational efforts in drug and biomarker development. This practice elevates research from being merely suggestive to being statistically robust and biologically authoritative.
In the era of data-intensive life sciences, multi-omics repositories serve as foundational pillars for biomedical discovery and therapeutic development. These repositories, such as The Cancer Genome Atlas (TCGA), Genotype-Tissue Expression (GTEx), and the European Nucleotide Archive (ENA), house vast quantities of genomic, transcriptomic, proteomic, and metabolomic data. A central dilemma for researchers utilizing these resources is the choice between accessing raw, primary data or pre-processed, analysis-ready datasets. This choice directly impacts the reproducibility, flexibility, and biological validity of downstream conclusions, particularly in high-stakes applications like biomarker identification and drug target validation. This whitepaper provides a technical assessment of both data formats, grounded in the practical realities of multi-omics research.
Defining Data Formats: Raw and Pre-processed
Raw Data refers to the primary, unaltered output from an analytical instrument. In multi-omics, this includes:
- Genomics: Binary Alignment Map (BAM) files from sequencers, containing read sequences and their alignment positions.
- Transcriptomics: FASTQ files with raw sequencing reads and quality scores.
- Proteomics:
.rawor.dfiles from mass spectrometers, with mass-to-charge ratios and intensity values. - Metabolomics: Proprietary instrument files containing chromatographic and spectral data.
Pre-processed Data has undergone a series of computational steps to transform raw signals into interpretable biological quantities. Common forms include:
- Genomics: Variant Call Format (VCF) files (mutations), or read count matrices (for RNA-seq).
- Transcriptomics: Fragments Per Kilobase Million (FPKM) or Transcripts Per Million (TPM) values in tab-delimited files.
- Proteomics: Peptide or protein abundance matrices, often normalized.
- Metabolomics: Peak area or concentration tables, with metabolite identifiers.
Benefits and Pitfalls: A Comparative Analysis
The following tables summarize the core advantages and disadvantages of each data format.
Table 1: Quantitative Comparison of Key Characteristics
| Characteristic | Raw Data | Pre-processed Data |
|---|---|---|
| Storage Volume | Very High (TB to PB scale) | Significantly Reduced (GB to TB scale) |
| Computational Demand | High (Requires HPC/cloud) | Low to Moderate (Often manageable on a workstation) |
| Reprocessing Frequency | Infrequent, resource-intensive | Common, as algorithms improve |
| Common Access Latency | Higher (often via controlled access) | Lower (often directly downloadable) |
| Format Standardization | Low (Instrument/center-specific) | High (Community-standard formats) |
| Metadata Complexity | High (Requires detailed experiment logs) | Moderate (Often curated) |
Table 2: Qualitative Benefits and Pitfalls
| Aspect | Benefits of Raw Data | Pitfalls of Raw Data | Benefits of Pre-processed Data | Pitfalls of Pre-processed Data |
|---|---|---|---|---|
| Analytical Flexibility | Unlimited. Can apply novel pipelines, adjust parameters, re-align, or extract novel signals. | None. | Limited to the choices embedded in the processing pipeline. | High. "Black-box" processing locks researchers into prior assumptions. |
| Reproducibility & Transparency | Enables full provenance tracking from machine output to result. | Requires exhaustive documentation of computational environment and code. | Simplifies replication if the same pipeline is used. | Irreproducible if processing software, version, or parameters are not fully disclosed. |
| Data Quality Control | Allows for sample-level, read-level, or peak-level QC. Enables filtering of low-quality data. | Requires significant bioinformatics expertise. | QC is typically performed, saving researcher time. | May mask underlying quality issues. Cannot rectify upstream technical artifacts. |
| Accessibility & Efficiency | Ideal for novel method development and deep, customized analysis. | Steep learning curve and infrastructure barrier. | Democratizes access for domain biologists. Accelerates hypothesis testing. | May be unsuitable for novel integrative analyses (e.g., splicing variants, post-translational modifications). |
| Comparative Analysis | Challenging due to batch effects and heterogeneous processing needs. | Standardized processing enables direct cross-study comparisons. | Hidden batch effects from the processing pipeline can confound biological signals. |
Experimental Protocols for Data Format Comparison
To empirically assess the impact of data format choice, researchers can conduct the following key experiments.
Protocol 1: Differential Expression Analysis Pipeline Comparison
- Objective: Quantify the variance in final gene lists introduced by using pre-processed counts vs. generating counts from raw reads.
- Methodology:
- Select an RNA-seq dataset (e.g., from GEO) with available raw FASTQ and pre-processed count matrix.
- Arm A (Raw): Process FASTQs through a modern alignment pipeline (e.g., STAR -> featureCounts). Apply standard normalization (e.g., DESeq2's median of ratios).
- Arm B (Pre-processed): Use the repository-provided gene count matrix directly.
- Perform differential expression analysis on both datasets using the same statistical model (e.g., DESeq2, edgeR).
- Compare the resulting lists of significant differentially expressed genes (DEGs) using Jaccard index and correlation of log2 fold changes.
Protocol 2: Variant Calling Concordance Study
- Objective: Evaluate the sensitivity and precision of variant calls from a repository VCF file vs. a re-analysis of BAM files.
- Methodology:
- Obtain matched tumor-normal whole-genome sequencing data (BAM files) and the associated VCF from a repository like TCGA.
- Arm A (Raw): Re-process BAMs through a GATK best-practices pipeline (HaplotypeCaller) or a modern deep-learning tool (e.g., DeepVariant).
- Arm B (Pre-processed): Use the repository VCF directly.
- Use a benchmark region (e.g., GIAB truth set) to calculate concordance metrics: Recall (Sensitivity), Precision, and F1-score for single-nucleotide variants (SNVs) and indels for each arm.
Visualizing the Data Processing and Decision Workflow
Data Processing Pipeline from Raw to Pre-processed
Decision Guide: Choosing Between Data Formats
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Tools for Multi-omics Data Analysis
| Tool/Resource | Category | Primary Function in Data Format Assessment |
|---|---|---|
| Galaxy Platform | Workflow Management | Provides accessible, reproducible pipelines for processing raw data (FASTQ to counts) without command-line expertise. |
| Nextflow/Snakemake | Workflow Orchestration | Enables scalable, portable, and reproducible execution of complex raw data processing pipelines on HPC/cloud. |
| Docker/Singularity | Containerization | Packages entire software environments (e.g., a specific GATK version) to guarantee processing reproducibility for raw data. |
| MultiQC | Quality Control | Aggregates QC reports from multiple tools and samples into a single HTML report, crucial for assessing raw data quality. |
| BioContainers | Software Repository | A registry of ready-to-use containers for bioinformatics tools, streamlining the setup for raw data analysis. |
| Jupyter/RStudio | Interactive Analysis | Environments for exploratory analysis and visualization of both raw data metrics and pre-processed data matrices. |
| Refinery Platform | Data Visualization | A tool for interactive exploration of large-scale pre-processed omics data from repositories like TCGA. |
| GEN3 | Data Commons Framework | Powers many modern repositories, providing APIs for querying and accessing both raw and processed data objects. |
The choice between pre-processed and raw data is not binary but strategic. For exploratory analysis, hypothesis generation, and educational purposes, high-quality pre-processed data from trusted repositories offers unparalleled efficiency. For novel algorithm development, deep mechanistic investigation, or when the latest processing methods significantly outperform those used in the repository, investing in the analysis of raw data is necessary. The future of multi-omics repositories lies in providing both formats alongside exhaustive, machine-readable metadata detailing every step of pre-processing. This dual approach, coupled with the tools and protocols outlined herein, will empower researchers to fully leverage the transformative potential of shared multi-omics data for precision medicine and drug discovery.
Within the context of multi-omics data repositories and resources research, selecting the appropriate data repository is a foundational step that directly impacts the reproducibility, accessibility, and long-term utility of scientific research. As data volumes and complexity grow, particularly in drug development, a systematic approach is required. This guide provides a technical checklist, framed by core criteria, to enable researchers, scientists, and professionals to make an informed choice.
Core Selection Criteria & Quantitative Data
The following criteria are distilled from current best practices and repository evaluations. Quantitative data is synthesized from recent analyses of major repositories.
Table 1: Quantitative Comparison of Major Multi-omics Repository Features
| Repository Name | Primary Data Types | Max Individual File Size | Accepted Formats | Embargo Support | Cost Model (Public Data) | DOI Minting | API Access |
|---|---|---|---|---|---|---|---|
| ArrayExpress | Transcriptomics | 50 GB | CEL, FASTQ, BAM | Yes | Free | Yes | REST, JSON |
| BioStudies | Multi-omics, general | 100 GB | Any | Yes | Free | Yes | REST |
| ENA (EMBL-EBI) | Genomics, Metagenomics | No stated limit | FASTQ, BAM, CRAM | Yes | Free | Yes | REST, Webin |
| GEO (NCBI) | Transcriptomics, Methylation | 50 GB (FTP) | SOFT, MINiML, RAW | Yes | Free | Yes | e-Utilities |
| MetaboLights | Metabolomics | 50 GB | mzML, nmrML | Yes | Free | Yes | REST, Java API |
| PRIDE (ProteomeXchange) | Proteomics, Mass Spectrometry | 50 GB | mzML, mzIdentML | Yes | Free | Yes | REST API |
| Synapse (Sage Bionetworks) | General, Clinical | 1 TB (via client) | Any | Yes | Free (quotas apply) | Yes | R/Python Clients, REST |
| Zenodo (CERN) | General, Supplementary | 50 GB | Any | Yes | Free | Yes | REST API |
Table 2: Qualitative Checklist for Repository Evaluation
| Criterion Category | Specific Question | Score (1-5) | Notes |
|---|---|---|---|
| 1. Scientific Scope & Suitability | Is the repository domain-specific (e.g., proteomics) or general? | Domain-specific repositories often offer better curation and tools. | |
| Does it mandate/use community-standard metadata schemas (e.g., MIAME, MIAPE)? | Critical for interoperability and reuse. | ||
| 2. Data Management & Curation | What is the level of provided curation (none, basic, enhanced)? | Enhanced curation adds significant value. | |
| Does it perform basic file validation and integrity checks? | Prevents deposition of corrupted data. | ||
| 3. Access & Sharing Policies | Are access controls granular (e.g., project-level, file-level)? | Essential for controlled-access or pre-publication data. | |
| What are the licensing options (CC0, CC-BY, custom)? | CC-BY is often required for journal compliance. | ||
| 4. Technical Infrastructure & Stability | What is the uptime/SLA guarantee (if any)? | Look for >99% uptime. | |
| Is the data stored in multiple geographic locations? | Ensures preservation against local failure. | ||
| 5. Long-term Preservation & Sustainability | Does it have a formal preservation plan (e.g., OAIS model)? | Indicates commitment to long-term data safety. | |
| What is the funding model (institutional, grant-based, fee-for-service)? | Stable funding reduces risk of repository sunsetting. | ||
| 6. Integration & Interoperability | Does it provide bi-directional links to relevant publications (PubMed IDs)? | Facilitates discovery. | |
| Is it integrated with major search portals (e.g., OmicsDI, Google Dataset Search)? | Increases data visibility. |
Detailed Methodologies: Repository Evaluation Protocol
To apply the checklist systematically, follow this experimental evaluation protocol.
Experimental Protocol 1: Metadata Completeness Assessment
- Objective: Quantify the adherence of a candidate repository to field-specific metadata standards.
- Materials:
- A prepared dataset from your project with corresponding metadata.
- The mandatory metadata submission template from the candidate repository.
- The relevant minimum information standard checklist (e.g., MIBBI portal resources).
- Procedure: a. Extraction: List all required fields in the repository's submission template. b. Mapping: Map each required field to the corresponding element in the formal community standard (e.g., MIAME for microarray data). c. Scoring: Assign a score: 2 = direct match, 1 = partial/indirect match, 0 = no match or missing critical field. d. Calculation: Calculate a "Metadata Compliance Score" as (Total Score / (2 * Number of Standard Fields)) * 100%.
- Expected Output: A percentage score. Repositories with scores >85% are considered to have strong standards alignment.
Experimental Protocol 2: Data Retrieval & Reusability Benchmark
- Objective: Measure the ease and efficiency of accessing and re-using data from the repository.
- Materials: A list of 10 known accession IDs (e.g., E-GEOD-XXXXX) for data similar to your target type. A standard computing environment with
curlor programming language (R/Python) installed. - Procedure: a. API Testing: For each accession ID, use the repository's public API to retrieve: (i) core metadata, (ii) the file manifest, (iii) a key file (e.g., a processed matrix). b. Timing: Record the time-to-first-byte and total download time for each operation. c. Scripting: Write a minimal script to automate steps a-b. Note the complexity (lines of code, need for authentication). d. Format Check: Verify that downloaded data files are in open, non-proprietary formats (e.g., BAM, mzML).
- Expected Output: A table of retrieval times and a qualitative assessment of API documentation and client library maturity.
Visualizations
Flowchart: Repository Selection Criteria Evaluation
Workflow: Data Deposition & Curation Process
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Tools for Repository Evaluation & Data Submission
| Tool / Reagent | Category | Primary Function | Example / Vendor |
|---|---|---|---|
| ISA (Investigation-Study-Assay) Framework | Metadata Standard | Provides a general-purpose, hierarchical metadata format to describe multi-omics experiments. | isa-tools.org |
| BioContainers / Docker | Software Environment | Ensures computational reproducibility by packaging analysis tools and pipelines into portable, executable containers. | biocontainers.pro |
| RO-Crate (Research Object Crate) | Packaging Standard | A method to package research data with its metadata and context into a single, reusable distribution format. | ro-crate.org |
| FAIRshake Toolkit | FAIR Assessment | Provides rubrics and APIs to manually or automatically assess the FAIRness (Findable, Accessible, Interoperable, Reusable) of digital resources. | fairshake.cloud |
| Webin Submission Tool | Data Submission CLI | The official command-line tool for high-volume or automated submissions to ENA, BioStudies, and MetaboLights. | EBI Webin |
| CyVerse Discovery Environment | Cloud Data Management | Provides a scalable platform for data storage, analysis, and sharing, often integrated with institutional repositories. | cyverse.org |
| DUST (Data Upload Support Tool) | Metadata Validator | A tool to validate spreadsheets of metadata against community-defined templates before repository submission. | EBI DUST |
Selecting the optimal repository is not merely an administrative task but a critical scientific decision that extends the lifecycle and impact of research data, particularly in multi-omics and drug development. By applying the systematic criteria, evaluation protocols, and tools outlined in this guide, researchers can ensure their data is deposited in a repository that maximizes its utility, ensures compliance with funder and publisher mandates, and contributes to the accelerating pace of open science. The "gold standard" is alignment with both project-specific needs and the broader ecosystem of FAIR data principles.
Conclusion
The expanding ecosystem of multi-omics repositories offers unprecedented opportunities for biomedical discovery and therapeutic development. Success hinges on moving beyond simple data retrieval to a strategic approach that encompasses thoughtful resource selection, robust integration methodologies, and rigorous validation. Future directions point toward even deeper integration of multi-omics with electronic health records (EHRs), real-time data sharing platforms, and AI-driven knowledge graphs that connect disparate data types. For researchers, mastering this landscape is no longer optional; it is a core competency essential for driving the next generation of translational, data-driven science. By leveraging the foundational resources, methodological tools, troubleshooting tactics, and validation frameworks outlined here, scientists can confidently navigate the multi-omics universe to generate robust, impactful, and clinically relevant insights.